eLife | 基于cfDNA甲基化和片段组学多模态分析,实现多癌种早检及定位
时间:2023-08-13 21:32:56 热度:37.1℃ 作者:网络
导读
全球范围内,癌症相关发病率和死亡率持续上升,高昂的治疗费用为多数癌症患者带来了巨大的经济负担。早期癌症检测可有效控制癌症进展、提高患者存活率以及降低医疗费用。
液体活检是一种非侵入性癌症早期检测方法,可捕获广泛的肿瘤特征,包括cfDNA、外泌体等。cfDNA是检测早期癌症的一种生物标志物,在患者的诊断、预后和治疗监测方面显示出明显优势。近年来,人们探索了cfDNA在癌症早期检测和起源组织定位方面的潜力,并开发了一系列检测方法。这些方法具有较大的潜力,但仍面临早期癌症患者血液中cfDNA含量低、测序深度较高等局限。
近日,越南国立癌症医院、医学遗传学研究所等机构的研究人员在eLife发表了题为“Multimodal analysis of methylomics and fragmentomics in plasma cell-free DNA for multi-cancer early detection and localization”的文章。研究团队开发了一种被称为“SPOT-MAS”的多模态方法,通过DNA甲基化及其大小检测肿瘤是否存在,全面分析cfDNA的甲基组学、片段组学、DNA拷贝数和末端基序,可从单一筛查中同时检测和定位癌症。经验证,SPOT-MAS能够以高灵敏度、高成本效益进行多癌症早期检测和肿瘤定位。SPOT-MAS有望成为一种普遍、简单、经济的方法,适用于大人群癌症早期检测。

文章发表在eLife
主要研究内容
癌症和健康参与者的临床特征
该研究共招募了5种常见癌症类型的738例患者以及1,550名健康参与者,包括乳腺癌、结直肠癌、胃癌、肝癌和肺癌。根据癌症类型,通过影像学和组织学分析对上述患者进行诊断,并随机分配到发现队列和验证队列中。
发现队列包括499名癌症患者和1,076名健康参与者,用于分析多个癌症和组织特异性签名并构建机器学习算法;验证队列包括239名癌症患者和474名健康参与者,用于对机器学习模型进行外部性能评估。
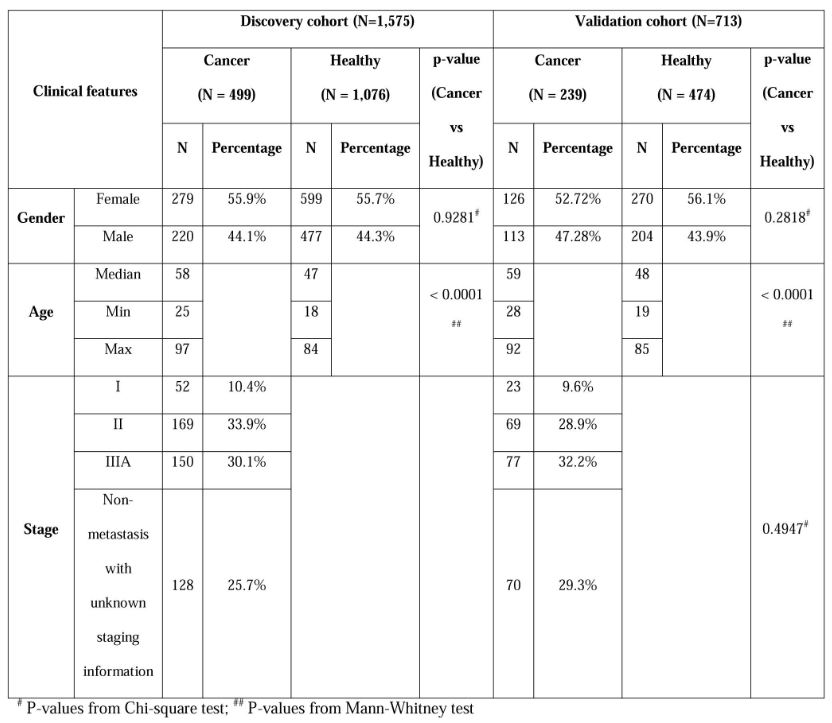
图1. 纳入队列信息汇总。
多模态SPOT-MAS检测方法
据介绍,SPOT-MAS具备一套新颖且高成本效益的工作流程,涉及三个主要步骤:第一步,从外周血中分离cfDNA,以构建cfDNA全基因组亚硫酸盐文库。第二步,使用特异性探针对文库进行杂交以收集靶标捕获部分,并通过全基因组测序(0.55X)和创新的机器学习算法进行分析;将所得数据预处理生成五组不同的cfDNA特征,包括靶区甲基化变化(TM)、全基因组甲基化(GWM)、片段长度模式(Flen)、拷贝数畸变(CNA)和末端基序(EM)。最后,采用机器学习模型和图卷积神经网络(GCNN)对癌症进行分类、识别TOO。
利用SPOT-MAS在发现、验证队列中进行分析,发现SPOT-MAS准确检测出了五种癌症类型,灵敏度为72.4%、特异性为97.0%。其中,乳腺癌检出率最低,肝癌检出率最高。
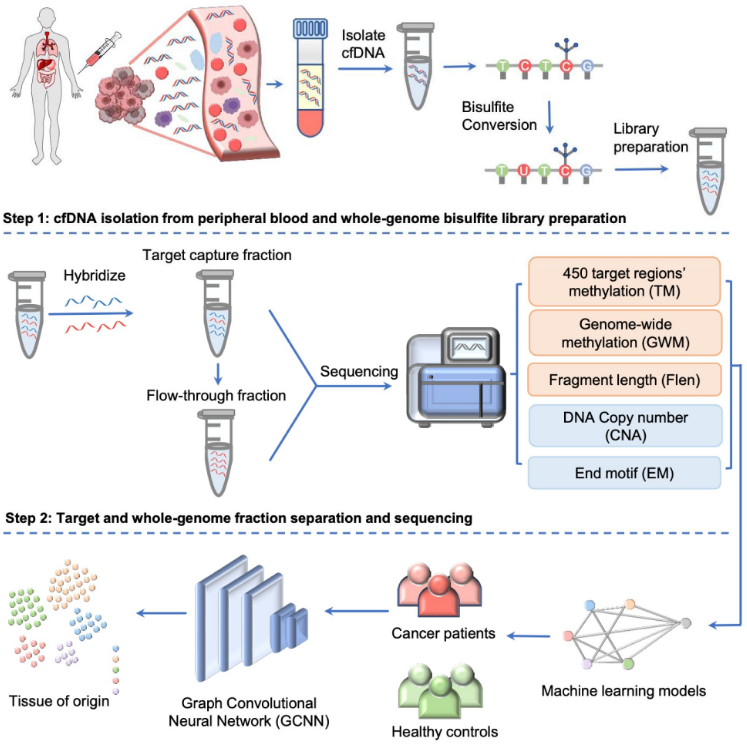
图2. SPOT-MAS工作流程。
从靶标捕获部分鉴定癌症患者的差异甲基化区
DNA甲基化是一个重要的表观遗传特征,可影响癌症相关基因表达。研究团队从公开数据中选择了450个癌症相关靶标基因区域进行分析。在癌症患者中共识别出402个显著的差异性甲基化区域(DMR),有339个区域被鉴定为高甲基化、63个区域被鉴定为低甲基化。
为了解DMR与生物通路之间的关系,研究团队对高甲基化DMR进行了通路富集分析,共检测到36条已知的重要富集通路,能够调节乳腺癌、胃癌、肝细胞癌和结直肠癌的发生。上述结果表明,靶标区域的甲基化变化,特别是高甲基化DMR,大多发生在肿瘤发生的早期,是区分早期癌症患者和健康个体的关键。
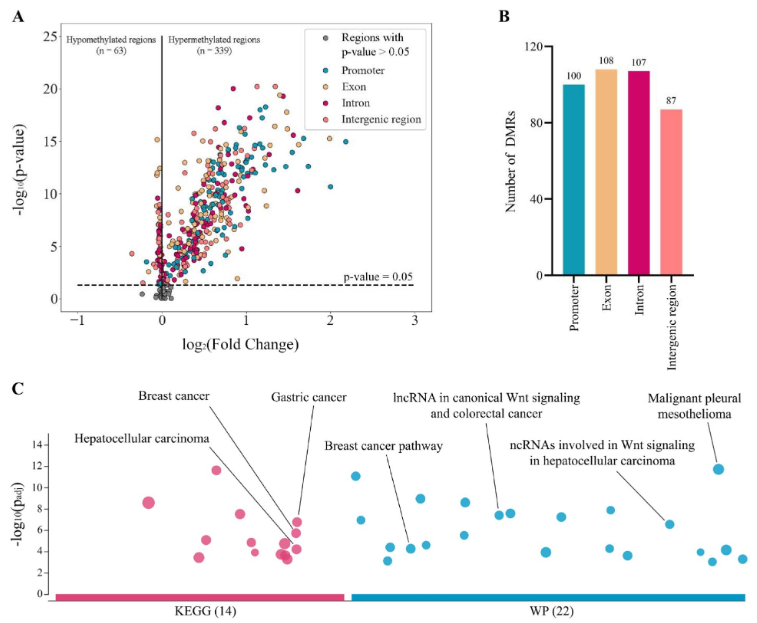
图3. cfDNA的靶向甲基化分析。
血浆ctDNA的片段长度分析
SPOT-MAS不仅可用于甲基化分析,还可用于片段大小分析。为检验癌症衍生cfDNA和非癌症衍生cfDNA的片段长度变化是否具有位置依赖性,研究团队通过SPOT-MAS计算了癌症患者和健康对照者基因组中短片段(≤150bp)与长片段(> 150bp)的比例。结果显示,癌症患者的cfDNA片段化程度高于健康参与者,其平均比值为0.29,健康参与组的平均比值0.27。此外,平均比值的变化分布在基因组的22个常染色体上。综上,SPOT-MAS技术可有效地捕获整个基因组中癌症和健康参与者之间片段化模式差异,使其成为检测血浆ctDNA的潜在生物标志物。
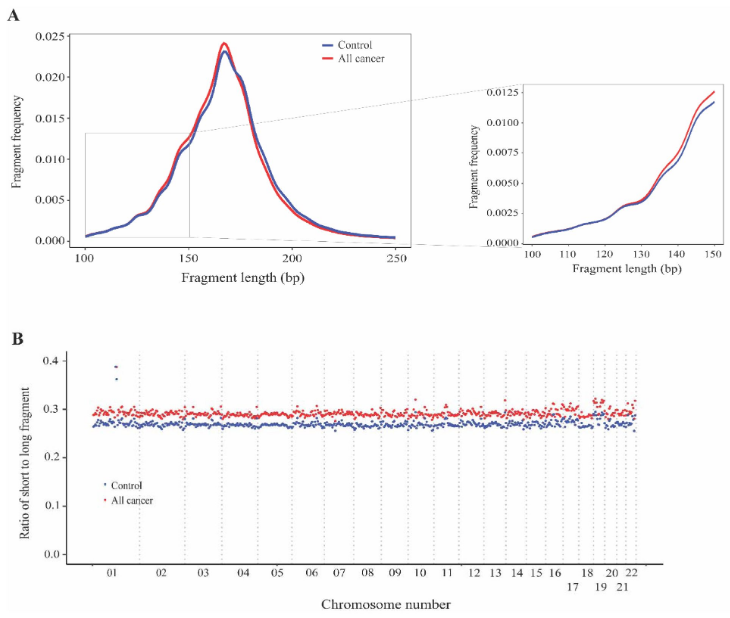
图4. 基于SPOT-MAS的cfDNA的片段长度分析。
SPOT-MAS检测可提高肿瘤检测的准确性
为提高早期癌症检测敏感性,同时避免高成本的深度测序,筛查检测应涉及广泛的ctDNA特征。因此,研究团队利用多个ctDNA特征来构建分类模型,以区分癌症患者和健康个体。为扩展特征空间,研究团队还基于片段长度生成了四个额外的特征,包括短片段计数、长片段计数、总片段计数和长/短片段比值,共计9个输入特征组。对于每个特征组,分别测试了随机森林(RF)、逻辑回归(LR)和极端梯度提升(XGB)三种不同的算法。结果显示,在9个特征模型中,基于EM模型的AUC最高,为0.90,基于短片段模型的AUC最低,为0.71。
接下来,研究团队探究了组合特征或集成模型能否改善分类,使用两种策略构建了多特征模型。结果显示,基于TM、GW、CNA、FLEN、LONG、TOTAL、RATIO和EM等7个特征的集成模型表现出最佳性能,AUC为93.95,优于单特征模型。

图5. 多特征集成模型可提高检测准确性。
SPOT-MAS能够预测癌症类型、识别起源组织
研究团队探究了SPOT-MAS识别癌症起源组织的能力,重点分析了发现队列中499名癌症患者,并结合9个cfDNA特征组构建了一个RF和两个神经网络(卷积神经网络和GCNN)模型。10倍交叉验证结果显示,基于GCNN的多特征模型在不同癌症中识别起源组织的性能最优,中位准确率为0.73(范围为0.54-0.87)。
在五种癌症类型中,乳腺癌(0.87)和肝癌(0.82)的起源组织识别准确率最高,胃癌(0.54)、结直肠癌(0.66)的准确率最低。上述结果强调了将多模态分析与GCNN模型相结合的益处,可捕获不同癌症类型中组织特异性标志物的广泛分布情况,有助于提高起源组织预测准确性。
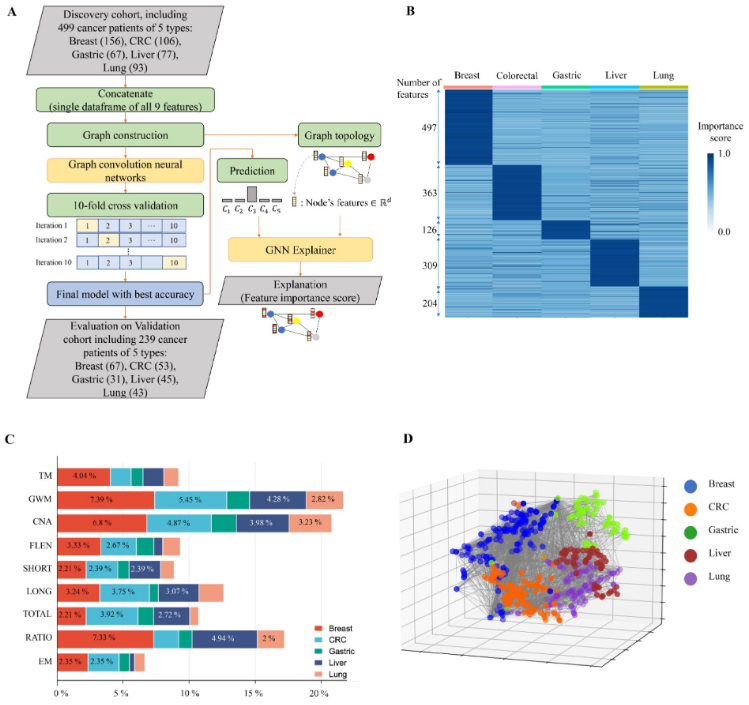
图6. SPOT-MAS在预测TOO方面的性能。
结语
综上所述,研究团队开发的SPOT-MAS检测方法可全面分析血浆cfDNA的多种生物标志物。大规模病例对照研究表明,SPOT-MAS将cfDNA特征的多模态分析和创新的机器学习算法相结合,能够低成本检测和定位多种癌症类型,为将SPOT-MAS作为高危人群的补充癌症筛查方法纳入临床环境提供了重要的支持。
参考文献:
1.Van Thien Chi Nguyen, Trong Hieu Nguyen, Nhu Nhat Tan Doan, et al. Multimodal analysis of methylomics and fragmentomics in plasma cell-free DNA for multi-cancer early detection and localization, eLife (2023).
https://elifesciences.org/reviewed-preprints/89083

