Alzheimer’s Dement:糖基化水平降低影响散发性阿尔茨海默病发病
时间:2023-05-09 10:43:51 热度:37.1℃ 作者:网络
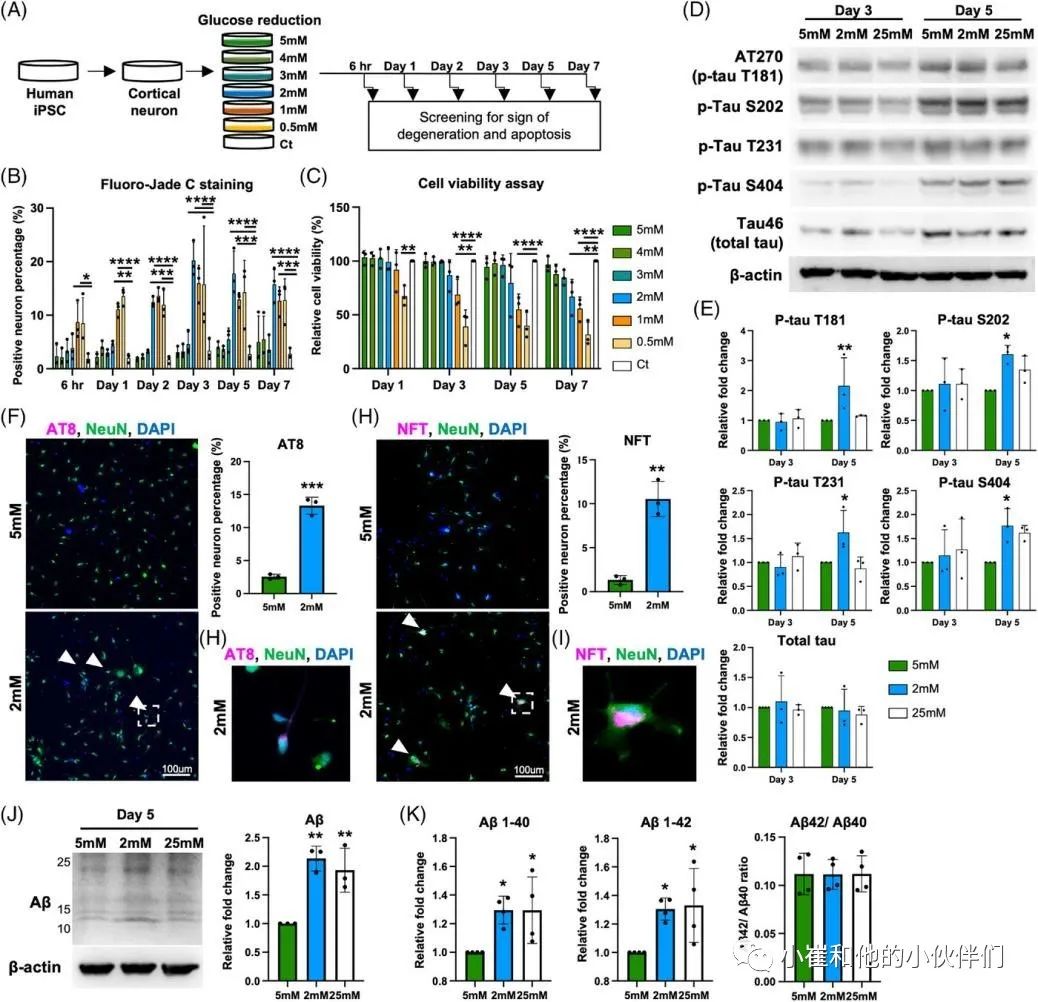
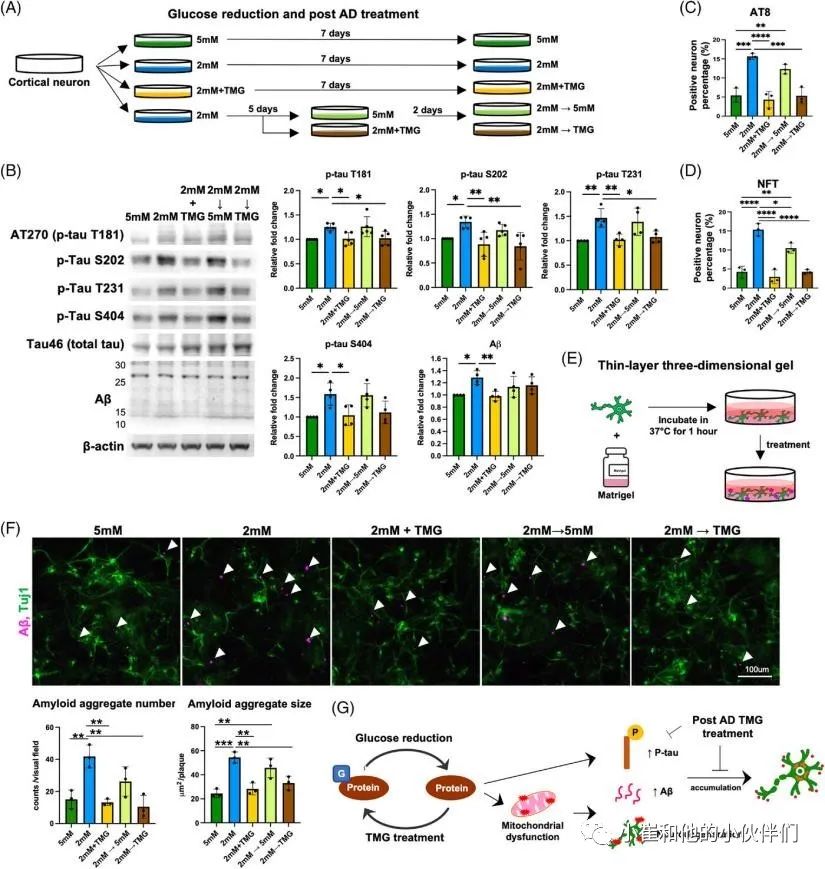
散发性阿尔茨海默病(sAD)是痴呆症的主要类型。O-GLcNAc糖基化是一种受调控的翻译后修饰,涉及在9000多种人类核蛋白、细胞质蛋白和线粒体蛋白的特定丝氨酸/苏氨酸残基上添加一个N-乙酰氨基葡萄糖(GlcNAc)残基。sAD患者大脑葡萄糖代谢低下,伴随着O-GLcNAc糖基化水平的降低,在症状出现之前就发生了,并与发病机制有关。到目前为止,由于缺乏人类sAD模型,O-GLcNAc糖基化在sAD病理中的作用和机制在很大程度上仍不清楚。 近期,Chia-Wei Huang等人在《Alzheimer’s Dement》期刊上发表了题为“Low glucose induced Alzheimer’s disease-like biochemical changes in human induced pluripotent stem cell-derived neurons is due to dysregulated O-GlcNAcylation”的文章。在作者团队的模型中,他们揭示了O-GlcNAc的失调,部分是通过线粒体功能障碍,导致了sAD样变化的发生,证明了抑制O-GlcNAcase在缓解AD样生化变化方面的治疗潜力。表明了O-GlcNAc的失调可能是低代谢和sAD样改变之间的直接分子联系。 为了使用人类神经元细胞研究散发性阿尔茨海默病(sAD),作者团队从健康人K3 iPSCs中产生并表征了混合皮质神经元,这些细胞没有已知的与AD相关的基因突变。鉴于葡萄糖摄取和代谢受损,以及有限的葡萄糖可获得性,可能参与了sAD的启动,作者团队假设慢性和中度葡萄糖缺乏可以诱导AD的病理。为了解决这个问题,他们将神经维持培养基替换为降糖培养基用于不同的时间段(图1A),降糖培养基是从具有0.5至5 mM范围内的较低葡萄糖浓度的神经维持培养基改造而来。每天更换培养基,并监测培养基中的葡萄糖浓度,以确保葡萄糖波动不超过0.5 mM。通过荧光-JADE C染色和细胞活力测定筛选了减少葡萄糖对成熟皮质神经元退化状态的影响(图1B,C)。将在含有25 mM葡萄糖的神经维持培养基中维持的神经元用作对照。将葡萄糖水平降低至1和0.5 mM分别诱导6小时内的神经元变性和72小时内的死亡,如Fluoro-Jade C染色和细胞活力测定所揭示的。另一方面,用高于3 mM的葡萄糖水平处理的神经元没有显示出任何退化或凋亡的迹象。有趣的是,2 mM的低糖处理在处理2天后导致变性神经元的急剧增加,并在第7天导致细胞活力降低。这些结果表明,由2 mM葡萄糖处理诱导的神经变性可能更好地代表在AD患者大脑中发生的慢性和中度葡萄糖缺乏下的AD样变化。因此,在接下来的研究中,使用2 mM的低糖处理来诱导神经变性,而用5 mM的葡萄糖浓度代替25 mM作为对照,这与生理葡萄糖浓度显著不同。 为了研究低糖诱导的神经变性是否与AD样特征相关,并可能作为研究sAD的模型,分析了两种主要的AD相关变化,包括tau磷酸化和Aβ水平。鉴于在低糖处理(2 mM)后的第3天和第5天出现最高百分比的变性神经元,在这些时间点收获用降糖培养基处理的神经元。通过蛋白质印迹法测量了与AD病理相关的几个磷酸化位点的p-tau蛋白水平(图1D)。与5 mM相比,2 mM葡萄糖处理的tau磷酸化水平在第3天没有变化。然而,它们在第5天显著升高,特别是在Thr181、Ser202、Thr231和Ser404上(图1D,E)。通过2 mM葡萄糖处理的p-tau水平的增加持续到处理的第7天,并在第10天开始消退,这可能是由于RIPA不溶性tau和Aβ部分的水平增加。tau蛋白的过度磷酸化是其毒性和聚集形成神经原纤维缠结(NFT)的原因,并且发现这些类型的tau蛋白在AD患者的大脑中积聚。因此,进行免疫荧光染色,分别使用AT8和NFT抗体检测过度磷酸化的成对螺旋丝(PHF)阳性tau蛋白和NFT阳性tau蛋白的沉积(图1F,H)。通过2 mM葡萄糖处理,AT8阳性神经元增加到13.3%(图1F),NFT阳性神经元增加到10.5%(图1H)。此外,NFT阳性神经元显示出气球样的表型(图1I ),而不是AT8阳性神经元(图1G)。结果可能表明AT8阳性神经元和NFT阳性神经元处于tau病理的不同阶段。接下来,作者团队使用与Aβ的N末端(氨基酸1-4)反应的MOAB-2抗体,评估了低糖处理对神经元内Aβ水平的影响。在作者团队的模型中检测到的分子量低于30 kDa的主要Aβ条带约为12、16、20、24和28 kDa,分别对应于三聚体(或C99)、四聚体、五聚体、六聚体和七聚体,因此是他们所定量的。在处理的第5天,与5 mM葡萄糖相比,2 mM葡萄糖显著诱导神经元内Aβ水平增高2.1倍(图1J)。通过多阵列电化学发光酶联免疫吸附试验(ELISA)进一步分析不同Aβ变体的细胞外水平(图1K)。将葡萄糖降至2 mM增加了Aβ1-40和Aβ1-42的水平,而没有改变Aβ1-42与Aβ1-40的比率,有趣的是,与5 mM葡萄糖处理相比,维持在恒定高葡萄糖(25 mM)的神经元没有改变p-tau水平,但诱导了皮质神经元中更高的Aβ水平(图1D–E,J–K)。这一观察结果与以前的研究一致,以前的研究表明高血糖会增加APP和Aβ水平。总的来说,这些数据表明,将葡萄糖降至2 mM可以诱导人皮质神经元中tau磷酸化和Aβ水平。 图1 将葡萄糖浓度降低到临界水平会在人类皮质神经元中诱导AD样表型 为了研究低葡萄糖诱导的 AD 样生化变化是否可以在源自其他干细胞系的皮层神经元中再现,皮层神经元是从另外两个健康人 PSC 系 C1 iPSC 和 H9 ESC 中产生的。C1 iPSC 和 H9 ESC 细胞系均可成功诱导分化为皮层神经元,神经元标记物和皮层谱系标记物的基因表达增加证实了这一点。2 mM 葡萄糖处理诱导来自 C1和 H9 系的皮层神经元中的 p-tau 和 Aβ 水平。最重要的是,在 2 mM 葡萄糖处理的神经元中也观察到 AT8 阳性 tau 和 NFT 阳性 tau 的积累增加。此外,AD 以不同和特定的方式影响大脑区域和神经元群。众所周知,小脑对 AD 病理具有相对抵抗力;因此,作者团队在小脑神经元上测试了他们的模型,以研究它是否概括了 AD 的可预测区域特异性方式。作者团队根据既定方案从人类 PSC 生成小脑神经元。使用浦肯野细胞标记物钙结合蛋白和颗粒细胞标记物 PAX6 通过免疫荧光染色确认小脑神经元的身份。混合小脑神经元用 5 或 2 mM 葡萄糖培养基处理 5 天,并通过蛋白质印迹检测 p-tau 和 Aβ 的水平。有趣的是,与皮质神经元不同,低葡萄糖处理不会改变小脑神经元中的 p-tau 或 Aβ 水平。综上所述,作者团队的结果表明,大约 2 mM 的葡萄糖浓度是诱导皮层神经元而非小脑神经元神经变性和 AD 样生化变化的关键水平,这些变化源自多种细胞系。 除了p-tau和Aβ病理学之外,轴突损伤和突触缺失是在sAD患者大脑中观察到的其他关键特征,它们主要与认知障碍相关。作者团队探索了轴突损伤和突触缺失是否可以在他们的模型中重现。为了评估降低葡萄糖浓度对神经突的影响,作者团队对用5 mM葡萄糖或2 mM低糖培养基处理5天的皮质神经元进行了Tuj1染色。结果显示,与5 mM葡萄糖处理的神经元相比,低葡萄糖处理的神经元中的轴突覆盖面积显著降低(图2A)。相应地,在2 mM处理的神经元中观察到轴突断裂,这是神经变性的标志。通过蛋白质印迹分析了低糖对突触的影响,结果显示低糖处理降低了synapsin-1的水平(图2B)。与蛋白质印迹结果一致,在低糖处理的神经元中通过免疫荧光染色也观察到突触密度的显著降低(图2C)。这些结果表明,作者团队目前的模型可以重现关键的AD样变化,不仅是p-tau和Aβ,而且还有轴突损伤和突触丢失。 多项研究表明AD患者的神经元功能发生了改变。因此,作者团队还通过多电极阵列(MEA)试验评估了低糖处理对神经元活动的影响。作者团队能够检测到代表处理前分化的皮质神经元中神经元成熟的指标的自发放电尖峰和爆发以及由同步指数指示的神经元网络活动。用不同浓度的葡萄糖处理后,每天测量神经元的活动。有趣的是,作者团队在第2天观察到2 mM低糖处理的神经元的神经元活动降低,随后活动逐渐增加,与用5 mM葡萄糖处理的神经元相比,在第5天后平均放电率增加达到2.5倍(图2D)。2 mM葡萄糖处理导致的平均放电率增加可能表明神经元过度兴奋,这是一种神经变性表型,在其他体外和体内AD样模型以及sAD初始阶段的患者大脑中均有报道。相比之下,指示突触功能的同步指数随着低糖处理导致的神经元活性改变而降低,并在第7天有显著差异(图2E)。这些结果共同表明,低糖暴露会损害人iPSC衍生的皮质神经元的神经元结构和功能。总之,该系统复制了AD的病理特征,包括p-tau积聚、Aβ增加和突触功能受损,并可作为一个平台来探索sAD中涉及的详细分子机制,特别是在sAD的早期阶段。 图2 将葡萄糖浓度降低到临界水平会改变神经元的结构和活动 在AD患者死后的大脑中观察到O-GlcNAc糖基化水平降低,并与AD的病理和进展相关。然而,O-GlcNAc在疾病起始和进展中的分子作用仍不清楚。作者团队采用新建立的模型来回答了这些问题。作者团队首先研究了在降低葡萄糖浓度后不同时间点的O-GlcNAc糖基化水平,通过使用一种抗体的蛋白质印迹法,该抗体在与蛋白质结合时特异性识别O-GlcNAc。作者团队发现低糖处理6小时后,O-GlcNAc糖基化水平显著降低(图3A)。尽管O-GlcNAc水平在低糖处理的12小时后反弹,但该水平再次显著下降并维持低水平直至实验结束(图3A)。除了O-GlcNAc糖基化水平,在处理的第5天,与正常葡萄糖培养基中的神经元相比,低糖处理也显著提高了O-GlcNAc转移酶(OGT)与O-GlcNAcase(OGA)的比率,这可能进一步表明O-GlcNAc体内平衡的变化。此外,第5天的样品通过二维凝胶电泳(2-DE)进一步分离,该电泳基于第一维的电荷和第二维的质量分离蛋白质。使用抗O-GlcNAc抗体检测O-GlcNAc,进行2-DE分析(图3B)。虽然通过SDS-PAGE和蛋白质印迹显示,通过低葡萄糖处理,O-GlcNAc水平整体降低(图3A),但2-DE提供了更多信息,即每个点上的O-GlcNAc糖基化受到不同的调节(图3B)。 接下来,为了验证O-GlcNAc糖基化在低葡萄糖诱导的AD样生化改变中的作用,一种有效的选择性OGA抑制剂Thiamet-G (TMG)被用于人工提高低葡萄糖处理的神经元中的O-GlcNAc糖基化水平。当使用浓度超过1μM时,TMG处理以剂量依赖的方式升高O-GlcNAc糖基化水平,并达到平台期(与赋形剂相比增加约4.5倍)(图3C)。此外,通过TMG处理,OGT表达水平和OGT/OGA比率降低,而OGA表达水平以剂量依赖性方式增加。为了更进一步,作者团队研究了增加O-GlcNAc糖基化水平是否能逆转低葡萄糖诱导的AD样生化变化。用低糖和不同浓度的TMG处理皮层神经元,用蛋白质印迹检测p-tau和Aβ水平。作者团队发现,通过抑制OGA,特别是在低糖处理的神经元中,提高O-GlcNAc糖基化水平,挽救了低糖诱导的AD样生化变化(图3D)。在不同浓度中,10nM的TMG处理在挽救低葡萄糖诱导的AD样生化变化方面最有效,而处理浓度高于10nM的神经元否定了有益的效果。这些数据表明在皮层神经元中精确调节提高O-GlcNAc糖基化水平的重要性。TMG对低糖处理的神经元的处理效果进一步得到AT8阳性tau(图3E)和NFT阳性tau(图3F)沉积减少,以及细胞外Aβ1-40和Aβ1-42水平降低(图3G)的支持。为了进一步证实在模型中观察到的低糖诱导的AD样改变是由O-GlcNAc糖基化减少引起的,一种小分子OGT抑制剂OSMI-4被用于降低神经元中的O-GlcNAc糖基化水平。用普通葡萄糖培养基(5 mM)和20nM或200nM的OSMI-4处理皮质神经元,通过蛋白质印迹测定p-tau和Aβ的变化。作者团队发现,5天的OSMI-4处理可以浓度依赖地诱导p-tau和Aβ水平。由于其他细胞途径也可能对葡萄糖缺乏做出反应,作者团队探索了是否有任何其他细胞途径,如AMPK和内质网(ER)应激,受到低糖处理的影响。在3、6、12、24、48和72小时后,在低糖处理的神经元中测量AMPK活化和ER应激的水平。有趣的是,在葡萄糖减少的情况下,AMPK和内质网应激水平都没有显著增加,这是由PERK和EIF2α的磷酸化水平决定的。这些结果表明,AMPK激活和内质网应激可能不涉及低糖诱导的AD样改变的早期阶段。总之,这些结果首次证明,在人类皮质神经元系统中,失调的O-GlcNAc糖基化是葡萄糖缺乏和AD样生化变化之间的分子联系,O-GlcNAc糖基化作为一种营养传感器调节无数细胞过程。大量神经元蛋白上的O-GlcNAc糖基化减少可能在AD样改变的启动中起关键作用。 图3 在低糖处理下,失调的O-GlcNAc糖基化在诱导AD样生化变化中起重要作用 令人信服的证据表明,氧化应激增加和线粒体功能障碍在AD病理的进展中起着重要的作用。此外,O-GlcNAc糖基化对参与调控线粒体功能许多方面的线粒体蛋白有深远的影响。因此,作者团队探索了低糖处理引起的O-GlcNAc糖基化失调是否会导致线粒体功能障碍,并进一步导致AD样生化变化。首先,为了阐明低糖处理是否影响线粒体糖基化水平,作者团队从细胞质中分离出线粒体,并通过蛋白质印迹分析这两个部位中的O-GlcNAc水平。作者团队观察到不仅在细胞质中而且在线粒体中O-GlcNAc糖基化水平显著降低(图4A)。接下来,为了评估低糖处理对线粒体健康和功能的影响,分别通过JC-1和CM-H2DCFDA染色检测膜电位和氧化应激。低糖处理在第1天线粒体膜电位出现了降低,并且在第2天后降低更显著(图4B)。膜电位的降低伴随着氧化应激的增加,从处理后第2天开始并持续到第5天(图4C)。这两种线粒体异常都可以通过10nM TMG处理使O-GlcNAc糖基化水平提高来挽救(图4B,D)。这些结果表明,低糖处理导致的O-GlcNAc糖基化水平降低与线粒体异常密切相关,并且可能是AD的最初原因之一。为了评估线粒体功能的关键参数,作者团队进一步采用海马XF细胞线粒体应激试验。皮质神经元用5或2 mM葡萄糖培养基处理,并在第2天和第5天分析线粒体功能(图4E–G)。测量了响应不同线粒体应激测试化合物的耗氧率(OCR)的变化(图4E和F)。作者团队观察到基础呼吸率和与ATP相关的呼吸率显著下降,这在处理的第5天变得更加显著,同时在低糖处理的第2天和第5天最大和备用呼吸能力减弱(图4G)。这些指标的下降表明呼吸链受损,这可能进一步导致生物能衰竭,使神经元易于退化。有趣的是,10nM的TMG处理在第2天部分恢复了低糖诱导的线粒体功能下降,并在处理的第5天进一步改善(图4E-G)。 为了进一步了解O-GlcNAc糖基化失调、线粒体功能受损和AD样生化变化之间的相互关系,作者团队研究了几种对不同AD病理特征表现出保护作用的化合物。J147通过靶向ATP合酶对与衰老相关的神经退行性疾病具有神经保护作用。3CHIR-99021是一种高度选择性的GSK-3β抑制剂,据报道可抑制tau磷酸化,而LY2886721是一种Aβ分泌酶抑制剂,可在临床试验中降低脑脊液中的Aβ水平。通过p-tau、Aβ和突触蛋白-1的表达来评估这些化合物的保护作用。用低糖以及TMG、J147、CHIR-99021或LY2886721处理皮质神经元,并使用蛋白质印迹法检测AD特征(图4H)。在低糖处理下,TMG处理显著降低了p-tau和Aβ水平,并恢复了突触蛋白-1水平(图4H)。令作者团队惊讶的是,J147处理对Aβ和突触蛋白-1水平的影响与TMG处理相似,但未能降低p-tau水平(图4H)。CHIR-99021和LY2886721处理分别如预期的那样显著降低了低葡萄糖诱导的p-tau和Aβ水平(图4H)。然而,通过CHIR-99021处理降低p-tau水平并不能降低Aβ水平,反之亦然。相应地,CHIR-99021或L Y2886721的处理未能恢复突触蛋白-1的水平。总之,这些结果表明,低糖诱导的AD样生化变化部分归因于线粒体功能障碍(图4I)。线粒体功能障碍似乎增加了Aβ水平并损害了突触,这是神经变性的标志,而O-GlcNAc糖基化的降低可以独立于线粒体异常增加tau磷酸化。有趣的是,作者团队的研究结果还表明,神经退行性表型、p-tau蛋白和Aβ蛋白可以平行观察到,彼此之间没有联系。总之,作者团队的模型概括了AD样改变的不同方面,这使得它不仅是研究AD分子机制的更有吸引力的平台,也是测试化合物治疗潜力的更有吸引力的平台。 图4 线粒体异常与O-GlcNAc糖基化水平降低密切相关,并先于AD样表型 由于作者团队已经证明了O-GlcNAc在sAD样改变的启动中的关键作用(图3),作者团队想知道在AD样改变发展后提高O-GlcNAc糖基化是否可以减缓进展并具有治疗效果。为了回答这个问题,对用低糖培养基处理了5天的皮质神经元应用了不同的处理(图5A)。经过5天的低糖处理后,将低糖培养基换成5 mM或含有TMG的低糖培养基,再进行2天。对于阴性和阳性对照,也收获用5 mM葡萄糖或2mM葡萄糖培养基处理的神经元,同时进行或不进行TMG处理。通过蛋白质印迹分析AD后TMG处理对减轻AD样生化变化(包括p-tau和Aβ水平)的有益作用(图5B)。TMG处理(2mM+TMG)降低了p-tau水平,AD后TMG处理(2mM→TMG)降低了较小程度,而2mM→5mM处理对于降低p-tau水平不明显。通过AT8和NFT染色证实了该结果,其显示AD后TMG处理(2mM→TMG)比5 mM葡萄糖培养基(2mM→5mM)对减少tau积聚具有更好的效果(AT8的图5C,NFT的图5D)。 然而,2mM→5mM和2mM→TMG处理都未能降低Aβ水平。因此,作者团队探索了延迟TMG处理是否改变了Aβ的沉积而不改变其产生。Aβ斑块的沉积是AD诊断的最重要指标之一,然而,在二维体外培养系统中很难观察到细胞外Aβ聚集。因此,作者团队开发了一种三维培养系统,其中皮质神经元与Matrigel混合形成薄层三维凝胶,允许Aβ肽在细胞外基质中沉积和聚集(图5E)。使用这种培养模型,能够评估低糖处理和TMG处理对Aβ聚集物形成的影响。 作者团队发现,与5 mM葡萄糖处理相比,低糖处理导致Aβ聚集体的数量和大小增加(图5F)。有趣的是,同时进行的TMG (2mM+TMG)和AD后TMG (2mM→TMG)处理可以挽救低糖诱导的Aβ聚集物的形成。改用5 mM培养基(2mM→5mM)仅略微降低了Aβ聚集的数量和大小。这些数据表明,同时进行TMG处理可以在早期阶段阻止导致AD样改变的级联反应,而对于AD后的特征,TMG处理可以通过减轻tau磷酸化和Aβ聚集形成来减缓进展(图5G)。总之,作者团队的结果支持使用OGA抑制剂作为治疗AD的方法来减缓进展和缓解症状。 图5 人工提高O-GlcNAc糖基化水平缓解p-tau和Aβ的进展 作者团队第一次表明,仅降低葡萄糖的可利用性就重现了人类皮质神经元的AD样特征。在该模型中观察到的表型与在sAD患者神经元中观察到的表型高度一致。通过这个模型,支持了这样一个假设,即大脑中葡萄糖代谢不足可能是使sAD直接产生淀粉样蛋白和tau蛋白畸变的潜在病因。作者团队假设,由葡萄糖缺乏引起的O-GlcNAc糖基化异常降低直接参与了sAD发病的早期阶段,并且与糖代谢受损和AD病理的发作有直接的分子联系。作者团队在这篇文章中也展示了一个人类神经元模型,该模型可以作为更好地理解AD病理的分子机制的工具,特别是在sAD的早期阶段,并且该系统也可以作为药物筛选的有力工具。 精读原文链接: Huang CW, Rust NC, Wu HF, Yin A, Zeltner N, Yin H, Hart GW. Low glucose induced Alzheimer's disease-like biochemical changes in human induced pluripotent stem cell-derived neurons is due to dysregulated O-GlcNAcylation. Alzheimers Dement. 2023 Apr 10. doi: 10.1002/alz.13058.