Nature:生命科学的变革:DeepMind人工智能可以预测大量蛋白质的结构
时间:2021-07-27 23:43:56 热度:37.1℃ 作者:网络
蛋白质是生命所必需的,了解其结构可以促进对其功能的机械理解。通过巨大的实验努力,已经确定了约100000种独特蛋白质的结构,但这只是数十亿已知蛋白质序列的一小部分。确定单个蛋白质结构所需的数月到数年的艰苦努力使结构覆盖率受到了限制。精确的计算方法需要解决这一差距,并使大规模的结构生物信息学。仅仅根据蛋白质的氨基酸序列预测蛋白质将采用的三维结构是“蛋白质折叠问题”的结构预测部分,多年来一直是一个重要的开放研究问题。尽管最近取得了一些进展,但现有的方法远远达不到原子的准确度,尤其是在没有同源结构的情况下。
Kathryn Tunyasuvunakool等进行了研究开发,提供了一种计算方法,可以定期预测蛋白质结构与原子的准确性,即使没有已知的类似的结构。

在具有挑战性的蛋白质结构预测关键评估(CASP14)中,验证了基于神经网络的模型AlphaFold的完全重新设计版本,证明在大多数情况下精度与实验相当,并且大大优于其他方法。AlphaFold最新版本的基础是一种新的机器学习方法,它将蛋白质结构的物理和生物学知识,利用多序列比对,整合到深度学习算法的设计中。
人类基因组拥有超过20000种蛋白质的指令。但只有大约三分之一的人通过实验确定了他们的三维结构。在许多情况下,这些结构只是部分已知。这款由谷歌在伦敦的姐妹公司DeepMind开发的一个名为AlphaFold的人工智能(AI)转化工具,已经预测了几乎整个人类蛋白质组的结构(生物体表达的蛋白质的完整补体)。此外,该工具还预测了各种其他生物体的几乎完整的蛋白质组,从小鼠和玉米(玉米)到疟原虫(见“折叠选项”)。通过一个公共数据库可以获得的35万多个蛋白质结构,其准确性各不相同。但是研究人员说,这种资源将在今年年底增长到1.3亿个结构,它有可能彻底改变生命科学。
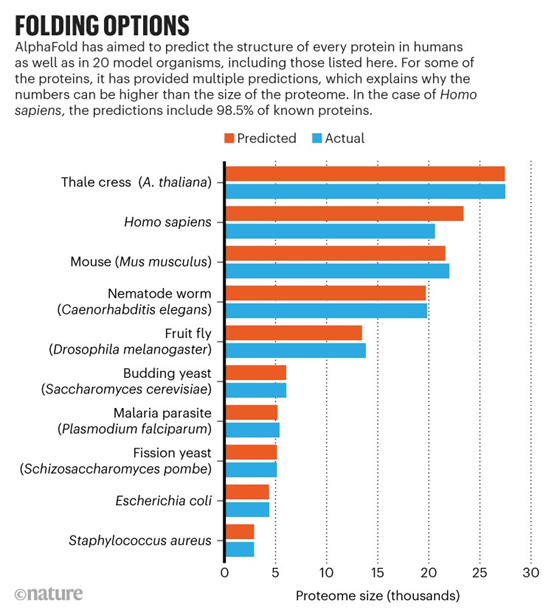
这完全是一场变革。这是迄今为止人工智能系统对提高科学知识做出的最大贡献。在准备公开发布AlphaFold代码的过程中,DeepMind对其进行了改进,使代码运行更高效。一些CASP预测需要几天时间,但是AlphaFold的更新版本现在可以在几分钟到几小时内计算出来。凭借这一更高的效率,DeepMind团队着手预测人类基因组编码的几乎所有已知蛋白质的结构,以及20种模式生物的结构。这些结构可在英国欣斯顿的EMBL-EBI(欧洲分子生物学实验室欧洲生物信息学研究所)维护的数据库中获得。AlphaFold预测的结构覆盖了98.5%的已知人类蛋白质,与其他生物的结构相似,此外,AlphaFold还对其预测的可信度进行了测量。
对于人类蛋白质组来说,58%的对单个氨基酸位置的预测足以确定蛋白质折叠的形状。这些预测中的一个子集——占总数的36%——可能足够精确,可以详细描述对药物设计有用的原子特征,比如酶的活性位点。即使是不太准确的预测也可能提供一些见解。生物学家认为,人类和其他真核生物(细胞有核的生物)的蛋白质中,有很大一部分含有固有无序的区域,只有在与其他分子协同作用的情况下才具有特定的结构。许多蛋白质只是在溶液中摆动,它们没有固定的结构。
AlphaFold预测的一些区域可信度较低,与生物学家怀疑的那些区域相符。确定单个蛋白质如何与其他细胞参与者相互作用是对AlphaFold预测的最大挑战之一。对于CASP竞争,它的大多数预测都是蛋白质的独立折叠单元,称为结构域。但是,人类和其他生物的蛋白质组中含有多个半独立折叠结构域的蛋白质。人类细胞也含有由多条相互作用的蛋白质链组成的分子,例如细胞膜上的受体。
随着新蛋白质的鉴定和预测的改进,数据库将会更新。研究人员已经在使用AlphaFold和相关工具来帮助理解X射线晶体学和低温电子显微镜产生的实验数据。有研究人员使用AlpHAFT从细菌的X射线数据中制作模型,用以躲避被称为粘菌素的抗生素。实验模型中与AlphaFold预测不同的部分通常是软件指定的置信度较低的区域,这表明AlphaFold正在准确预测其极限。
原文出处
Tunyasuvunakool, K., Adler, J., Wu, Z. et al. Highly accurate protein structure prediction for the human proteome. Nature (2021). https://doi.org/10.1038/s41586-021-03828-1

