Pharmacol Ther:ADC类药物临床开发中的经验教训
时间:2022-10-14 05:59:06 热度:37.1℃ 作者:网络
抗体偶联药物(ADC)是由靶向特异性抗原的单克隆抗体与小分子细胞毒性药物通过连接子链接而成,兼具传统小分子化疗的强大杀伤效应及抗体药物的肿瘤靶向性。ADCs于20世纪90年代中期首次进入临床试验以来,经过近30年的发展,已经成为一个非常成功的肿瘤学平台。
截止2021年12月,美国食品药品监督管理局(FDA)已批准了14个ADC药物,相比于基于抗体的新型免疫治疗靶点相比,在过去几年中批准了更多具有新靶点的ADC(TF、Nectin-4、CD19、Trop-2、BCMA、CD79b等)。然而,目前正在开发的ADC仍面临许多挑战,这些挑战以惊人的频率出现,很大程度上阻碍了临床的进展。
基于这一点,本文总结了ADC临床试验中获得的成功和失败的经验。以为未来更多ADC药物的开发获得更快、更安全的途径。
试验方案设计中的注意事项
选择合适的受试者
在第一阶段研究中,有太多的设计是为了选择更可能有反应的近乎完美的患者,而忽略了第一阶段确定肿瘤患者群体安全剂量的既定目标。事实上,资格限制性太强,以至于对研究患者的寻找变得缓慢,而所得结果却通常不适用于真正有需要的典型患者。因此,合格的选择标准应反映出需要临床试验的“真实”患者。
应该允许一些器官功能障碍患者
进入第一阶段研究的人群通常有难治性和晚期疾病的物理和生化症状。此外,这些癌症患者通常为老年人,也会患有高血压、糖尿病和慢性阻塞性肺病等与某些器官功能障碍相关的疾病。
如果临床前毒理学未显示肝或肾毒性,研究方案应允许这些器官出现一定程度的功能障碍。通常建议血清肌酐水平高达并包括正常上限(ULN)的1.5倍,肝转氨酶高达ULN的3倍,无肝转移。对于肝转移患者,建议肝转移患者使用5倍ULN。
不要排除碱性磷酸酶升高的患者,因为碱性磷酸酶可能来源于骨骼或肝脏,如果胆红素和肝转氨酶可以接受,则对耐受性几乎没有实际影响。由于前列腺癌、肺癌和乳腺癌患者的骨转移率高,碱性磷酸酶升高,可能不必要地将其排除在外。
不要根据血清白蛋白水平排除患者,因为没有证据表明白蛋白水平2.8比3.0在临床研究中更有意义。由于白蛋白水平与肿瘤负荷呈负相关,因此可能会无意中选择肿瘤负荷较小的患者。这一限制将导致重大偏差。
避免没有依据的特殊标准
一项研究武断地排除了在室内空气中血氧饱和度低于93%的患者。该标准没有规定它是处于休息状态还是处于活动状态。尽管其基本原理是降低发生严重肺毒性的风险,但该分子或有效载荷的肺损伤临床前毒理学研究没有数据。此外,没有已知证据表明研究前的血氧饱和度可以预测ADC的肺毒性。然而,这一排除标准肯定排除了许多肺癌患者、潜在慢性阻塞性肺病患者和肺转移患者。
应该允许使用抗凝药物和一定程度的凝血参数异常。血栓事件在许多癌症适应症中很常见,使用因子Xa抑制剂可以很好地控制和预防血栓事件。除非血小板减少症被预测为所选定ADC相关的毒性,否则将患者限制在不使用抗凝剂或需要正常凝血参数,将排除掉一些优秀的候选者。
此外,绝对淋巴细胞计数(ALC)下降是晚期癌症患者的常见表现。这一实验室发现对大多数患者的临床影响很小,限制ALC低的患者进入研究通常没有好处。反对限制ALC的最有力证据是pembrolizumab的首次临床试验。即使在这种非常成功的免疫治疗中,也没有使用ALC标准,因此不应将其纳入ADC开发方案中。
不要排除先前的ADC治疗者
这种筛选标准在一些较新的方案中已经观察到,这既不符合当代药物开发标准,也越来越不现实,因为近年来,许多ADC已经获得批准或正在临床开发中。
通常选择排除已有ADC治疗的理由是,通过排除先前接触过ADC的患者,可以降低对最新ADC产生获得性耐药性的风险。然而,患者对ADC没有反应的原因是多因素的,包括患者接受的剂量水平太低、肿瘤中目标抗原表达水平太低、连接子不稳定或肿瘤对有效载荷具有固有抗药性。此外,排除接受相同有效载荷的患者可能并不完全有效,因为在过去具有相似ADC有效载荷的ADC中观察到患者偶尔出现反应。
新治疗必须解决患者未满足的需求,而最有价值且最快的批准途径是基于先前治疗失败的患者的获益。加速批准trastuzumab deruxtecan(Enhertu:registered:)治疗HER2阳性转移性乳腺癌就是一个很好的例子。该研究特别包括了经trastuzumab-emtansine(Kadcyla:registered:)治疗失败的HER2阳性乳腺癌患者。显著的响应率以及延长的响应时间导致了加速批准。如果trastuzumab deruxtecan的研究排除了先前的ADC治疗,那么其很可能需要通过与trastuzumab emtansines进行比较的随机试验而获得常规批准。
找到正确的剂量
MTD通常来源于第一阶段试验的安全数据。但实际上,药物只有经过几个疗程后,累积毒性才会显现出来,其中有一些会表现的越来越严重。例如,一些有前景的ADC与仅在多个周期后发生的危及生命的肺炎有关。因此,建议剂量应在仔细分析多个患者、多个疗程以及剂量减少和剂量延迟后确定。在第一阶段花一些额外的时间完善推荐剂量,比必须回头然后再公开宣布在研究中减少剂量要好得多。
此外,许多临床试验的问题在于,主要研究者只是名义上的,患者由一系列研究员、住院医生和副研究者进行评估。这些人可能对常见毒性标准(NCI CTC)熟悉程度不同。看护患者的工作人员往往没有查阅与毒性等级相关的具体定义,而是使用了一种常见的任意描述,即轻度(1级)、中度(2级)、重度(3级)和危及生命(4级),来描述血液学和化学特征以外的毒性。这可能会导致药物毒性,尤其是非血液毒性的报告不足。拥有非常熟悉NCI CTC和对毒性分类具有丰富经验的研究人员,是准确收集安全数据和成功确定推荐剂量的关键。
最后,有证据表明,由于基于体重计算的剂量,毒性会反复发生。绝大多数ADC的剂量都是mg/kg或mg/m2,然而在肿瘤学中,这方面的科学证据很少,更不用说ADC了。基本药理学原理告诉我们,抗体或其他大分子治疗药物最初仅限于血管室。然而,由于脂肪组织没有良好的血管化,血管体积并没有与总体重成比例增加。肥胖或超重患者的基于体重的剂量增加,会使血浆浓度不成比例地增加,从而导致毒性增加。随着西方世界肥胖症的增加,这对研究对象来说是一个现实,必须考虑到临床试验中。此外,一些患者可能出现第三腔积液(腹水、水肿和胸腔积液)。
在这方面,mirvetuximab soravtansine是一个很好的例子。在该药物的早期临床开发中,剂量限制性角膜炎是一种非靶向肿瘤不良事件,当药物剂量为mg/kg时,在较高剂量下发生的频率较高。尽管做出了许多努力,但这种角膜毒性无法通过润滑或类固醇眼药水等局部措施缓解。这对后期开发的可行性提出了挑战。随后,研究者进行了详细的药代动力学分析和建模确定了Cmax与角膜炎严重程度之间的关系。进一步的研究表明,需要使用调整后的理想体重公式(AIBW)给药,以在一系列患者中实现安全有效的血浆浓度范围。这种给药方法在更广泛的卵巢癌患者群体中得到了进一步验证,在保持抗肿瘤活性的同时,角膜炎从40%显著减少到10%。
由于某些ADC的治疗指数小于非ADC抗体治疗,因此应仔细分析剂量、剂量公式、临床药代动力学和毒性之间的关系,并确定最佳剂量方法。
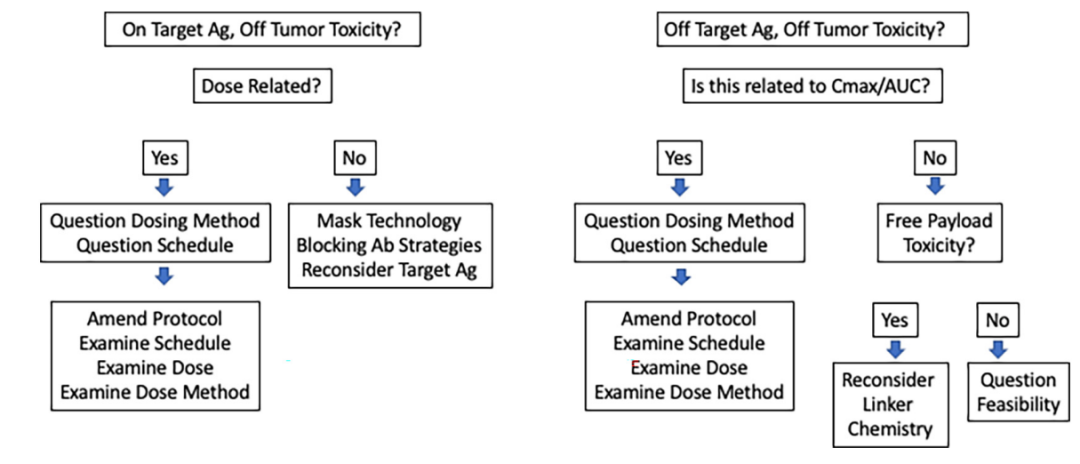 探索多种给药方案
探索多种给药方案
通常,ADC的第一阶段研究只选择一种给药方案。新药的常规给药计划通常来源于细胞毒性药物,每3周选择一次,因为这将接近血液学恢复时间。在抗体时代,剂量越来越基于预期的清除率和半衰期,导致每2至4周的给药计划。因此,对于ADC药物,没有理由采用细胞毒性药物或抗体的时间表。也许更好的选择是根据耐受性、药代动力学和/或靶抗原转换和靶抗原再生动力学选择时间表。
Gemtuzumab ozogamicin(Myelotarg:registered:)是ADC开发中极好的研究案例。Gemtuzumab ozogamicin是FDA批准的第一种ADC药物,于2000年批准,随后于2010年自愿退出,然后于2017年重新批准。该ADC的初始计划为每2周9mg/m2。在最早的研究中,由于血浆中药物浓度超过CD33+饱和水平的不成比例增加,观察到饱和药代动力学模式,剂量与血浆浓度呈非线性关系。这种药物在血浆中的积聚,高于使CD33+靶细胞饱和所必需的水平,至少在一定程度上是观察到的许多毒性的原因,包括骨髓抑制、急性肝毒性和移植后的肝静脉闭塞性疾病。2010年,在随后的验证性试验未能验证其临床疗效并证明其安全性, Gemtuzumab ozogamicin自愿退出市场。
后期的研究确定,AML靶细胞上CD33表达转换的动力学大约为每3-4天一次。随后,研究者提供了一种新的方案,即每3天给药3mg/m2的低剂量,给药3次。使用分次给药方案将Gemtuzumab ozogamicin的峰值和血浆总浓度降低至预期的剂量比例范围内。临床上,这产生了更大的耐受性和抗肿瘤活性,2017年,FDA再次批准了Gemtuzumab ozogamicin。
生物标志物的策略
ADC生物标记物开发的首要策略是识别并选择对ADC反应可能性最大的癌症患者亚群,从而实现高比率和持久的反应,确保监管部门的批准。实现这一战略的策略通常是生物标记物开发中最大的问题。
在早期研究中,尽可能接受可能表达靶点的肿瘤患者
对于刚刚描述的总体策略来说,这似乎与直觉背道而驰,但这是早期开发中最常见的错误之一,可能会显著降低临床执行速度。为了更好地了解目标抗原存在的预筛选问题,必须了解临床研究中组织采集和分析的真实过程。
许多患者没有易于获取的存档组织。而为了获得用于试验筛选的组织,需要一个漫长的过程,即使是进行分析的最快周转时间,通常仍需要6周才能获得结果。现在想象一下,患者患有晚期恶性肿瘤,对标准治疗无效,正在考虑进行临床试验。他们会等待预筛选结果吗?还是他们会进行另一个不需要预筛选的临床试验?答案对大多数人来说是显而易见的,并且反映在临床试验中心进入患者的方式上。
不要等到Assay完美
生物标记物的开发很复杂。尽管ADC有抗体作为骨架,但选择用于治疗的抗体可能无法令人满意地用于免疫组化(IHC)。为IHC生物标记物选择不同的抗体候选是非常常见的。通常的建议是,临床试验和生物标记物同步开发常常是最有效的。
不要迷信靶抗原的表达水平
必须承认,肿瘤细胞必须靶抗原,才能发挥ADC的抗肿瘤活性。然而,即使高表达也不能一定保证抗肿瘤活性,而低表达也不保证一定没有活性。ADC可能由于许多独立于肿瘤细胞表达的因素而无法引起肿瘤细胞死亡。
与直觉相反,一些在最初诊断时靶抗原低表达的患者观察到了反应。这可能是由于组织样本的年龄、质量和检测的特异性,也可能是由于随着疾病的进展,目标抗原表达增加。因此,早期开发中最合理的方法是不仅仅依靠早期临床试验中已证实的目标抗原表达来选择患者,还要利用扩张期的数据来确定表达和反应之间的关系。
此外,并非每个ADC都需要生物标记物来选择患者。对于许多血液肿瘤来说,靶细胞是一个特定的谱系,因此ADC可以靶向正常和恶性细胞,这方面的例子包括CD33+、CD22、CD79b和CD30。
最后,如果生物标记物不必要或任意地使患者无法接受潜在的有益治疗,这可能会限制以后的商业机会。
在研究中不要更改Assay或截止参数
这听起来可能不言而喻,但最近的经验表明情况并非如此。这种错误最近的一个例子是mirvetuximab soravtansine,一种针对铂难治性卵巢癌叶酸受体α(FRa)的ADC。在第二阶段研究中,使用有利于FRa高表达的选择标准,在铂类难治性卵巢癌中观察到有希望的抗肿瘤活性。
然而不幸的是,研究中改变了检测的患者选择参数,该研究包括具有中等FRa表达的患者。这导致无法达到无进展生存率(PFS)改善的主要终点。事后分析发现,高表达(FRa)患者的子集与早期研究中的患者一样,具有统计学意义。
ADC的标准临床药理学考虑
直到最近,ADC开发过程中监管批准所需的许多临床药理学步骤都没有与药物或生物制剂分开概述,这就提出了一个问题,ADC应该被视为细胞毒性药物,还是更接近抗体?这种不确定性导致人们质疑是否需要进行器官功能障碍研究、药物相互作用研究和全面的QTc研究。
最近,FDA药物评价与研究中心(CDER)和生物制品评价研究中心(CBER)联合发布了指南草案,以协助ADC的临床开发。这些非常实用的建议应该有助于行业研究人员确定需要进行哪些临床药理学研究。也可以说,指南的发布进一步证明了ADC是自身的平台,超越了细胞毒性和抗体开发。
小结
目前有超过20多种新型的ADC药物处于早期开发阶段,还有更多处于后期临床前研究阶段,ADC疗法改善癌症患者的前景非常看好。成功的关键往往是向他人学习并避免他人的错误。希望这篇文章可以提供一些有用和实用的方法,以确保临床开发的成功,更紧密地反映患者群体,这些经验教训将提供最佳的前进道路。
参考文献:
1.Antibody drug conjugates: The dos and don'ts in clinical development. Pharmacol Ther.2022 Jun 20;240:108235.

