Cell:JAK/STAT信号通路的前世今生
时间:2022-10-27 06:00:20 热度:37.1℃ 作者:网络
Janus 激酶 / 信号转导和转录激活子(JAK/STAT)信号通路是一种普遍表达的细胞内信号转导通路,参与细胞增殖、分化、凋亡和免疫调节等生物学过程。因此,JAK是药物研发的热门靶点。2022年10月13日,美国国立卫生研究院关节炎和肌肉骨骼和皮肤病研究所的John J.O’Shea博士为通讯作者,在JAK-STAT通路发现30年之际,在Cell杂志(IF=66.85)上非常有心地发表了一篇题为“The JAK-STAT pathway at 30: Much learned, much more to do”的综述文章,回顾了JAK/STAT在过去30年的里程碑发现和进展,并讨论了该领域目前面临的挑战及机遇。

摘要
JAK/STAT通路的发现源于对细胞如何响应干扰素 (IFN) 的研究,该通路揭示了从细菌到哺乳动物的细胞信号传导范例。关于该通路的相关发现揭示了由多种细胞外多肽(包括细胞因子、白细胞介素和相关因子)介导的基因表达机制。这些发现解释了许多人类疾病的发生发展机制,从免疫缺陷到癌症,并为治疗自身免疫、过敏和传染病的治疗提供理论依据,包括 COVID-19。尽管取得了这些进展,但仍然存在重大挑战和机遇。
简介
30年前,Janus 激酶 (JAK) 信号转导和转录激活因子 (STAT) 通路被发现。在这篇综述中,作者总结了该通路的发现历史及功能特点(图1)。对该通路研究的结果,让研究者了解细胞外多肽驱动生理和病理反应的机制,包括细胞和有机体发育、增殖、新陈代谢、感染、炎症和癌症。同时,大量研究获得的证据表明,该信号通路可影响基因组,诱导和抑制基因,这使得研究者考虑如何利用这些研究成果来开发针对该通路的调节剂。JAK-STAT 通路诞生以来,有大量研究结果发表。在这篇综述中,限于篇幅,作者只罗列并总结了具有影响力的研究成果。迄今为止,尽管在JAK-STAT通路研究方面取得了非凡的进步,仍然有需要解决及深度研究的问题。

图1:JAK-STAT通路研究历史。该时间表显示了自 1984 年以来JAK-STAT 通路研究的各种里程碑事件。GAS,伽马激活序列;IFN,干扰素;ISGs,干扰素刺激的基因;ISRE,干扰素敏感反应元件;SCID,严重联合免疫缺陷;SLE,系统性红斑狼疮;PK,假激酶;MPN,骨髓增生性肿瘤;GOF,功能增益;SOCS,细胞因子信号抑制因子;PIAS,活化 STAT 的蛋白质抑制剂;Tum-l,肿瘤致死。
JAK-STAT通路研究中的里程碑事件

JAK-STAT通路研究历史中的标志性事件。图片源自于Stark G R, Darnell Jr J E. The JAK-STAT Pathway at Twenty[J]. Immunity, 2012, 36(4); 503-514. Doi: 10.1016/j.immuni.2012.03.013
干扰素 (IFN) 于 1957 年被发现,可以迅速诱导机体抗病毒反应。JAK-STAT 通路的发现源于对细胞如何对 IFN 作出反应的研究。1980 年代末和 1990 年代初的前基因组时代,研究人员面临的挑战是阐明基因表达如何影响各种外源信号的机制。使用生物化学和分子和细胞生物学工具,研究者发现了G 蛋白偶联受体和受体酪氨酸激酶 (TYK) 的受体介导的信号转导,同时鉴定了特定的 DNA 结合蛋白和一系列转录因子。
1990年,Krebs 和 Fischer 使用相对较新的简并聚合酶链反应方法生成蛋白激酶文库,发现可逆的蛋白质磷酸化,并鉴定了TYK 家族,其中包括 TYK2。1991年和1992年,Harpur和Wilks应用相同的方法,发现了JAK1 和 JAK2。JAK 这个名字来源于罗马的双面神 Janus,代表这个激酶家族具有特征性的双面结构,其氨基末端激酶结构域前面是调节性假激酶结构域。然而,受限于当时的研究条件,JAK 激酶家族的功能仍然不是很清楚。
2012年,为了为庆祝JAK-STAT通路发现20周,Stark and Darnell在《Immunity》杂志上发表综述性文章,总结了JAK-STAT通路的发现历程,因此,对于研究历程,本综述在这里只做一个简短的摘要。通过众多研究者的努力,干扰素变为一种容易获得的试剂。1986年,Levy从 IFN 处理诱导的 mRNA 中产生 cDNA 文库中,发现几个 IFN 诱导的基因,确定了控制其表达的启动子和调控元件。1991年,Stark 和Kerr使用 IFN 诱导基因的启动子来驱动一种致命标记物的表达,该标记物可杀死所有对 IFN 有反应的细胞。1992年,Velazquez证实,该标记经过广泛的诱变,可分离出抗性细胞克隆,并通过基因互补,产生TYK2。Darnell, Stark,和Kerr随后证实,该标记存在于细胞质中,并迅速移动到细胞核,刺激细胞产生IFN。Darnell首次纯化提取了第一个 STAT(STAT1 和 STAT2,以及干扰素调节剂)因子 9 (IRF9),并确定它们的部分氨基酸序列。IRF9的功能具有双重性,并具有快速反应性。选择 IFN 作为初始研究目标,极大地促进了 JAK-STAT 通路的发现。因为IFN可诱导JAK-STAT 通路相关基因迅速表达,与调节细胞生长和分化的基因表达相比,抗病毒基因的表达模式与其有显著不同。
JAK-STAT通路详解
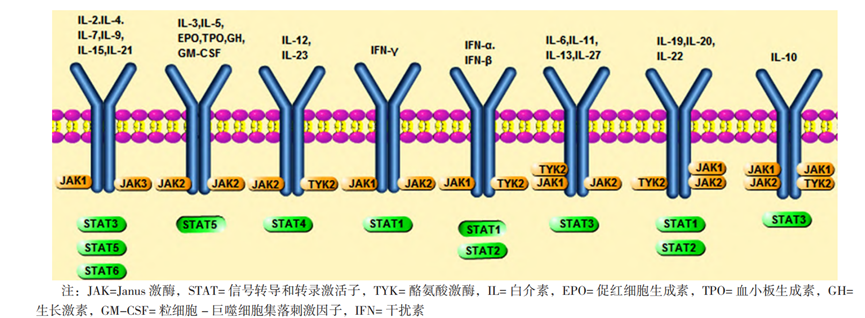
与 JAK/STAT 信号通路相关的多种细胞因子。图片源自于肖凡妮,青玉凤,张全波 . JAK/STAT 信号通路在风湿免疫性疾病中作用的研究进展[J]. 中国全科医学,2022,25(17):2159-2164. DOI:10.12114/j.issn.1007-9572.2022.0275.
Stark、Kerr和Darnell利用一组缺乏I型和II型IFN信号的细胞系,通过遗传互补,迅速定义了JAK-STAT信号通路的大致轮廓。JAK-STAT通路被许多细胞因子、生长因子和其他配体用来调节靶细胞中的基因表达。目前已知,近60种细胞因子,包括许多白细胞介素、集落刺激因子(CSFs)、激素样细胞因子和生长因子,参与JAK-STAT通路的调控。
随着基因组计划的完成,根据其独特的结构,JAK家族中有4个成员。最终鉴定出的成员是JAK3,与其他家族成员不同,它可以选择性表达(图2)。可激活JAK3 的细胞因子具有共同的 γ 链 (γc)。

图2:JAK-STAT信号通路。细胞因子与受体结合后,将信号传递给酪氨酸,酪氨酸磷酸化并激活受体相关的JAK,随后JAK磷酸化并激活STATs。酪氨酸磷酸化的STATs(pSTATs)形成二聚体,易位到细胞核,与目标DNA序列结合,并调节基因表达(pSTAT典型途径)。未磷酸化的STATs也会形成二聚体,进入细胞核,并调节转录(uSTAT非典型途径)。有多种诱导负反馈系统来抑制信号传导,包括细胞因子信号抑制因子(SOCS)家族蛋白、活化STAT(PIAS)家族蛋白抑制蛋白、蛋白酪氨酸磷酸酶(PTP)、USP18和ISG15。导致导致功能获得和/或功能丧失表型的人类单基因突变,每个组成基因的颜色编码为免疫缺陷(蓝色)、炎症(粉红色)或肿瘤(橙色)。
JAK 末端包含一个羧基末端催化或激酶结构域,前端是一个假激酶 (PK) 或激酶样结构域。JAK 通过氨基末端ezrin 、radixin、moesin (FERM) 结构域和 Src Homology 2 (SH2) 结构域,与细胞因子受体的Box1 和 Box2 结构域结合(图 3)。获得全长 JAK 的详细结构非常具有挑战性,直到2021年,才获得与细胞因子受体的细胞内结构域复合的全长 JAK1 结构(图 3)。JAK1 的结构域形成了一个扩展的结构单元,该结构单元可通过包装来自单体受体/激酶复合物的 PK 结构域,促进细胞因子受体/JAK 复合物的二聚化。

图 3:Janus激酶的结构。活化的Janus激酶二聚体(PDB:7T6F)与成熟的iFNlR1/IL-10Rb(紫色)及IFN-l(橙色)(PDB:5T5W)的胞内结构域结合。值得注意的是,该受体的细胞膜内部分可以通过n端Box1和c端Box2基序,与JAK1 FERM和SH2结构域结合。JAK激酶样或假激酶结构域,可以促进细胞因子受体/JAK复合物的二聚化。
STAT3 被认为是响应 IL-6 的 DNA 结合“急性期反应因子”,其行为类似于 IFN 激活的 STAT1,并且与 STAT1 的二聚体具有相似结构。值得注意的是,表皮生长因子和其他生长因子是 STAT3 激活剂,但受体 TYK 激活 STAT 不需要 JAK参与。在淋巴组织中富含STAT4,该分子可被 IL-12、IL-23 及 I 型 IFN 激活。STAT5是一种由催乳素激活的“乳腺特异性核因子”,包含两种同源基因:STAT5A和STAT5B。STAT5可被IL-2、IL-3、粒细胞巨噬细胞集落刺激因子(GM-CSF)、IL- 5激活。SATA6是IL-4 的诱导因子。
JAK激活结构域-细胞因子受体结合,导致受体尾部酪氨酸残基的 JAK 自身/转磷酸化、活化和磷酸化。受体的磷酸化,为细胞质的STATs 创造了对接位点,这些 STATs 通过酪氨酸磷酸结合 SH2 结构域,被募集到受体复合物中并自身被磷酸化。活化的磷酸化 STATs (pSTATs) 通过分子间 SH2-磷酸酪氨酸相互作用,形成同源或异源二聚化,转移到细胞核并结合 DNA 调节元件,改变染色质的可及性并诱导基因转录;然而,未磷酸化的 STATS (U-STATs) 也聚集在细胞核中。尽管通常可以针对给定的细胞因子,鉴定出主要作用的 STAT。但据一些研究结果,在某些情况下,一些细胞因子几乎可以激活所有 STAT,并且所有 STAT 都具有激活大多数细胞因子的功能。
JAK-STAT 通路在动物中广泛存在,从秀丽隐杆线虫、黑腹果蝇到脊椎动物。果蝇具有三种相关的细胞因子样配体——Upd1、Upd2 和 Upd3、一种细胞因子样受体Domeless (Dome) ,均可导致JAK 的激活,Domeless又称为 Hopscotch (Hop)。
尽管 JAK-STAT 通路是细胞因子信号传导的主要通路,但该通路也会招募其他 SH2 信号分子,并且 细胞因子受也可以参与JAK-STAT之外通路的调控。这些分子通路包括 Ras、MAP 激酶、AKT1、PI3K 和 MTOR 通路。IL-2 刺激细胞的蛋白质组学分析结果显示,在IL-2 激活的 T 细胞中, 90% 的磷酸化蛋白质组是 JAK 依赖性的,其余的由 SRC 家族激酶信号介导,包括磷脂酰肌醇的产生和 AKT 活性。
JAK-STAT通路负向调控
1、生理性负向调控
与许多生物调控系统一样,负反馈系统是由正作用刺激诱导的,而 STAT 靶基因的细胞因子信号传导抑制因子(SOCS 家族)就是一个突出的例子。人类基因组中编码了八种 SOCS 蛋白,包括 SOCS1-7 和 CIS,它们包含一个中央 SH2 结构域和一个羧基末端 SOCS 盒。SOCS 蛋白与 STAT 竞争结合细胞因子受体,并通过 elongin BC 和 E3 连接酶 Cullin5 诱导其泛素化和降解来干扰细胞因子信号传导。SOCS1 和 SOCS3 包含一个称为激酶抑制区 (KIR) 的短基序,它直接与 JAK(JAK2、JAK1 和 TYK2)相互作用。SOCS1 KIR 通过阻碍 ATP 和底物的进入来抑制 JAK2,而 SOCS3 与 gp130 相关的 JAK2 相互作用。SOCS3 优先由 IL-6 诱导,而 SOCS1 主要抑制 IFN,但也抑制 IL-12、IL-4/13 和 IL-2 细胞因子家族。
其他关键的负反馈调节剂还有:泛素特异性肽酶 18 (USP18) 和 IFN 刺激基因 15 (ISG15)(图 2)。ISG15 是一种泛素样蛋白,可与靶蛋白共价偶联(ISGylation),调节多种宿主和病毒蛋白的功能,USP18 是一种蛋白酶,可将 ISG15 从靶蛋白中解偶联。两者均由 IFN 诱导,并且是 I 型 IFN 信号传导的关键负调节剂(图 2)。USP18 与 JAK1 竞争结合IFNAR2,而 STAT2 可以将 USP18 募集到 IFNAR2。此外,ISG15 与 USP18 非共价结合,防止其泛素化/降解。
负调节剂也可以靶向调节JAK-STAT 通路的下游(图 2)。激活的 STAT (PIAS) 家族蛋白质抑制剂包含多个成员,它们作用于 STAT 和其他因子,包括 NF-κB,通过多种机制干扰信号传导,例如阻断 TF 的 DNA 结合活性、招募转录共阻遏物和促进蛋白质糖化。据报道,多种磷酸酶促进 STATs 去磷酸化,包括 CD45、PTP1B、TC-PTP、PHLPP1、PTPRT、PRPRK、PTPN9 和 PTPN2。值得注意的是,SHP-1 和 SHP-2 对JAK-STAT 通路信号传导既有正面影响,也有负面影响。编码负向调节因子的基因具有保守保守序列,例如,在果蝇中,编码名为 Socs36E 的 SOCS 蛋白和名为dPIAS的单个 PIAS 蛋白的基因具有保守序列。
2、病毒相关的负向调控
直接或间接针对 STAT 的病毒抗免疫策略,突出了 STAT 介导的 IFN 信号作为抗病毒防御的重要性。STAT 介导的 IFN 拮抗机制包括靶向 STAT 降解、隔离和细胞质滞留。在许多类型病毒的感染中, STAT活性在宿主细胞中被抑制,以逃避 IFN 抗病毒反应。副粘病毒非结构蛋白(V、C 和 W)直接与STAT结合,抑制STAT活性。副流感病毒 (PIV) PIV5 、PIV2 以及腮腺炎病毒的 V 蛋白,与STAT1 或 STAT2 的泛素连接酶结合,以诱导蛋白酶体降解,从而抑制STAT活性。黄病毒,包括登革热病毒和寨卡病毒,靶向作用于 STAT2,促进STAT蛋白酶体依赖性降解。
Henipaviruses、Nipah 病毒和 Hendra 病毒以及麻疹病毒的V 蛋白与 STAT1 和 STAT2结合,防止STAT寡聚化、细胞溶质聚集体中的隔离、核转位和输入。二聚体 STAT1输入细胞核,与名为 importin 5的结合位点结合,而埃博拉病毒 VP24 蛋白特异性地阻断了这种相互结合。在肠道病毒和轮状病毒中,也发现了核转运蛋白对 STAT 核输入的破坏。SARS-CoV-2 ORF6 蛋白通过 Nup98-Rae1 核孔亚复合物干扰 STAT 输入。
其他病毒可以通过隔离在细胞溶质聚集体,来调节 STATs 或促进核输出。SARS-CoV-2 N 蛋白可介导 STAT 胞浆滞留, Henipavirus V 和 Chikungunya 病毒可以通过 CRM-1 ,主动将STATs输出细胞核。
STAT 结构、复合物形成和对功能的影响
所有 STAT 共享相似的 N 端、卷曲螺旋、DNA 结合、接头、SH2 和反式激活结构域 (AD)(图 4)。活化的 JAK 磷酸化具有一个 STAT 单体的保守酪氨酸残基,然后与另一个单体的 SH2 结构域结合,驱动构象变化,使 STAT 易位进入细胞核,启动靶基因的转录。在这篇综述中,作者使用 P-STAT 来指定“典型”酪氨酸磷酸化(即 STAT1-Y701、STAT2-Y690、STAT3-Y705、STAT4-Y693、STAT5-Y694 和 SYAY6-Y641)和 U-STAT 来指定没有酪氨酸磷酸化的 STAT。其他残基可能会或可能不会被磷酸化。

图4:STAT结构域。STAT蛋白包含以下结构域:n端结构域(ND)、螺旋-螺旋结构域(CCD)、DNA结合结构域(DBD)、连接结构域(LK)、Src同源2结构域(SH2)和反转录激活结构域(AD)(上面板)。关键的磷酸化酪氨酸和丝氨酸氨基酸位于AD中,用于STAT二聚化和功能调节。以上是STAT1二聚体的结构,特别是它们的未磷酸化的反平行构象(左)(PDB:1YVL)和与DNA结合的磷酸化的平行构象(右)(PDB:1BF5)。值得注意的是,反平行构象阻止了DBD通过与相反的二聚体的CCD的相互作用与DNA相互作用。与STAT1一样,STAT3(PDB:1BG1)和STAT6(PBD:4Y5W)具有与DNA结合的平行结构,而STAT3(PBD:6TLC)和STAT5a(PDB:1Y1U)具有STAT二聚体的反平行构象。
STATs 可以形成多种异源性或同源性二聚体。STAT1、STAT3 和 STAT5a 已显示以平行或反平行构象形成同源二聚体(图 4;以 STAT1 为例)。两个 P-STAT 的 DNA 结合结构域 (DBD) 形成平行构象的“ C ”形结构,与 DNA 结合。然而,在反平行同源二聚体中,DBD 通过其卷曲螺旋结构域 (CCD) 相互相互作用,阻止了其与 DNA 的结合。酪氨酸磷酸化调节这两种构象之间的平衡。例如,P-STAT1 主要是平行的,U-STAT1 主要是反平行的,但也存在低水平的替代构象。值得注意的是,U-STAT3、U-STAT5 和 U-STAT6 在细胞核中也具有调节基因表达或异染色质形成的功能。U-STAT3 与 IκB 竞争结合NF-κB ,然后在 importin-α3 的帮助下,将 U-STAT3/p65/p50 复合物带到细胞核,与 κB 位点结合,促进CCL5 基因的表达。U-STAT5 不是启动转录因子,而是作为一种肿瘤抑制因子发挥作用。它能够通过促进异染色质的形成,抑制多种癌基因。与 p300 相关的 U-STAT6 ,直接与 COX-2 γ 激活序列 (GAS) 结合,驱动其在非小细胞肺癌细胞中的表达。
N 端结构域 (ND) 对于调节 STAT 同源二聚体的形成、功能至关重要。当 P-STAT1 与 DNA 脱离时,ND 二聚化,有助于将磷酸化的酪氨酸暴露为 TC45 磷酸酶的靶标。此外,STAT 四聚体可以通过 ND 之间的相互作用,协同募集成对的同源二聚体形成。这种四聚体与结合位点的相邻 DNA 基因座结合。因此,与二聚体相比,四聚体形成更稳定的 DNA 复合物,并与 DNA 结合的更为牢固。四聚体的形成,对于 STAT1 对 II 型 IFN 的完全转录反应是必不可少的。对于 STAT5,四聚体的形成不仅直接诱导基因表达,而且间接调节基因抑制,这一过程可能是通过中间抑制因子的正向调节。
与形成同源二聚体的其他 STAT 不同,响应 IFN I 和 III 的基因表达由 STAT1:STAT2 异源性二聚体驱动,该异源性二聚体与 IRF9 一起,构成干扰素刺激的基因因子 3 (ISGF3)(图 5)。在 P-ISGF3 中,P-STAT1:P-STAT2 二聚体形成平行构象,通过与磷酸酪氨酸-SH2 相互作用而稳定构象平衡。在这种构象中,两个 STAT 的 DBD 与 IRF9 的 DBD 同时与 DNA 结合。在 U-ISGF3 中,两个 DBD可以协同结合 DNA。STATs 和 IRFs 之间的模块化关联,可以促成非经典复合物的形成,包括 IRF9 与未磷酸化的 STAT1 或 STAT2 的变异复合物。最近来自电子显微镜和生化分析的研究结构表明,U-STAT1 和 U-STAT2 异源性二聚体具有非常稳定的反平行构象,U-STAT1:U-STAT2 和 U-STAT2:IRF9 二聚体是互斥的。也就是说,IRF9 不能与 U-STAT1:U-STAT2 二聚体结合,U-STAT1 不能与 U-STAT2:IRF9 结合。根据这些崭新的研究结构,以前称为“U-ISGF3”的功能复合体。现在主要是 U-STAT2:IRF9 二聚体,U-STAT1 扮演着更外围的角色。在未受刺激的细胞中,U-STAT2:IRF9 复合物驱动大多数 ISG 的表达,并在 IFN-I 或 IFN-γ 刺激下,转换为包含 STAT1、STAT2 和 IRF9 的 ISGF3 复合物。

图5:STAT1和STAT2配合物的构象变化。STAT结构域的定义见图4。从结构上看,STAT1同型二聚体和STAT1:STAT2异型二聚体各自适应两种构象,称为平行和反平行。平行二聚体通过SH2结构域和磷酸化酪氨酸残基之间的相互作用来稳定,而反平行二聚体则通过ND相互作用来稳定。除了酪氨酸磷酸化外,两个新发现的STAT2的T387和T404磷酸化可调节U-STAT1:U-STAT2异质二聚体的稳定性。
此外,与 U-STAT2:IRF9 结合的功能性 IFN 刺激调节元件 (ISRE) ,可以存在于 NF-κB 驱动基因子集的调节元件上游区域,例如IL6 。重要的是,需要高水平的 U-STAT2 和 IRF9 来驱动基因表达。U-STAT1:U-STAT2 二聚体还以不依赖 IFN-I 的方式广泛发挥作用,抑制多种信号通路中的 STAT1 功能,例如 IFN-γ 和 IL-27。在这个 U-二聚体中,STAT2 通过两个 N 末端之间的相互作用,与STAT1 结合。由于 U-STAT2 主要位于未受刺激细胞的细胞质中,它在多个信号通路中抑制 STAT1 的磷酸化和核转位。
U-STAT1:U-STAT2 二聚体不仅抑制多种 P-STAT1 同型二聚体功能,而且抑制 ISGF3 形成。虽然IFN-I 的刺激会驱动 P-STAT2 形成,但调节 U-STAT2 功能的关键分子事件才刚刚被揭示,包括最近发现的 STAT2上的 T387 和 T404 ,这是两个重要的磷酸化位点(图5)。T387 的磷酸化有利于 U-STAT1:U-STAT2 异二聚体的形成,但是却抑制了酪氨酸磷酸化的异二聚体形成和 ISGF3活性。另一方面,T404 被 IKKi 或 TBK1 磷酸化,可以破坏稳定的 U-STAT1:U-STAT2 二聚体,促进对 IFN-I 的反应,并显着增加 ISGF3 对 ISRE 的亲和力。T404 的磷酸化也可以促进U-STAT2 与 STING1 的结合,抑制了 STING 介导 IFN-I 的产生。U-STAT2 的新功能机制以及 U-STAT2 在生物学中所起的作用需要更深入的研究去了解。
复合物的形成也会影响 STAT 的核质穿梭。核质穿梭通过核孔复合物发生,并通过与核转运蛋白-β 家族的转运蛋白(即输入蛋白和输出蛋白)的相互作用进行调节。STAT 的核运输并不总是依赖于酪氨酸磷酸化,不同的 STAT 之间,核运输的调节机制是不同的。STAT1 和 STAT2 在细胞质和细胞核之间的动态分布与其结合 DNA 的能力相关。STAT1 同源性二聚体的核转运依赖于酪氨酸磷酸化,其中 importin-α5 识别 P-STAT1 二聚体中的条件核定位信号 (NLS)是关键步骤。IRF9相关的NLS 促进了 U-STAT2:IRF9 复合物的核定位。此外,STAT2 的核输出与其磷酸化状态无关,而是由其羧基末端的核输出信号 (NES) 介导。STAT3 NLS 在其 CCD 中,STAT3 的核定位不依赖于酪氨酸磷酸化,但依赖于 importin-α3 的识别。对 STAT 的核运输的调节机制较为复杂,还需要更深入的研究去探索。
STATs 和转录调控
一旦 STAT 复合物到达细胞核,它们就会完成与转录调控相关的任务:参与染色质化的靶基因、结合反应元件,以及募集可以募集和激活 RNA 聚合酶 II (RNA Pol II) 的转录共激活因子。
1、ISG 的 STAT 调控
宿主细胞对病毒感染具有快速反应性,ISG 转录的诱导通常很快。这在ISG 转录中所出现的一些现象得以印证,包括增加的转录速率、快速mRNA 加工和降解速率以及没有转录后控制(即 microRNA、翻译)。IFN 刺激 ISG从头招募 RNA Pol II ,这与TATA 结合蛋白的 TAF II 130/GCN5 转录复合物相关。pTEFb 通过 BRD4, 促进ISG C-端磷酸化,从而导致 RNA Pol II 相关阻遏物负延伸因子和 DRB 敏感性诱导因子的释放,使 ISG mRNA 延伸成为可能,这一过程与 RNA Pol II 募集相协调。STAT2 AD 通过 MED14 和 MED17 亚基,使得STAT2 能够激活 RNA Pol II。
此外,在经过IFN 处理后,ISGF3 结合位点或附近的启动子和基因体的染色质可及性增加,促进SWI/SNF 核小体重塑复合亚基的募集。BRG1 是 SWI/SNF 核小体重塑复合物(或哺乳动物 BAF/pBAF 复合物)的 ATP 酶亚基,与 STAT2 相互作用,促进转录。RVB1 和 RVB2,SRCAP、TIP60、URI 和 INO80 组成染色质重塑复合物,该复合物与 STAT2活性相关。干扰 RVB1 ,会阻止 RNA Pol II 向 ISG 启动子募集,但确切的作用机制尚不清楚。此外,染色质重塑剂可以影响组蛋白乙酰化,导致 ISG 启动子的组蛋白 H4 乙酰化降低。同样,乙酰化染色质可以增强溴结构域的募集,促进BRG1与ISGF3的结合。
H2A.Z 核小体在 ISG 转录中发挥调节作用,shRNA 对 H2A.Z 的干扰可增加 ISGF3 活性,导致ISG mRNA 的产生增加,从而更有效地抑制病毒复制。相应地,ISGF3 可以诱导 H2A.Z 脱离启动子,组蛋白乙酰转移酶 (HAT) GCN5 或溴结构域蛋白 BRD2 的抑制会破坏 这一过程。值得注意的是, CREB 结合蛋白(CBP、组蛋白去乙酰化酶 (HDACs) 和 RVB1对H2A.Z并无影响,但ISG 转录在H2A.Z 脱离启动子这一过程中是必不可少的。近期一项研究证实,组蛋白 H3. 3 也在 ISG 转录中发挥调节作用,具体机制与H2A.Z机制类似。
在SG 转录激活过程中,组蛋白乙酰化和去乙酰化具有重要意义,多个控制位点有助于基于乙酰化的 IFN/细胞因子信号传导。多个STAT家族成员 与 HAT 相关,例如,STAT2 与 CBP/p300 和 GCN5 相关,而 p300 与 STAT1、STAT3、STAT5 和 STAT6 相关。干扰 GCN5 或 CBP 会抑制 ISG 转录,但从 ISG 启动子中驱逐 H2A.Z 取决于 GCN5活性,而不是 CBP活性,这证明了 HAT 功能具有特异性,并且具有多种调节功能。同样,ISG 转录可以被 HDAC 蛋白和 HDAC 抑制剂所抑制,因为 HDAC 抑制剂可以阻止RNA Pol II 向 ISG 启动子的募集。此外,HDAC 抑制可能使得BRD4 结构域乙酰化,从而限制其对 STAT 的激活。由于 BRD4 在高乙酰化和低乙酰化染色质上的可逆隔离,BRD4 和 HDAC 对 ISG 调节的可能会实现交叉调节。
2、STATs 和其他转录因子
在生理条件下,细胞会同时接收多个活化因子,从而激活多个信号通路。典型的例子是 Toll 样受体 (TLR) 和细胞因子信号(如 IFN-γ),它们分别激活 NF-κB 和 JAK-STAT 通路。STAT 与其他 TF 之间相互作用,已有大量文献研究成果,包括调节 TF 合成、调节 TF 活性以及基因组内影响转录的直接或间接相互作用。最值得注意的是,免疫反应中的一些关键基因,需要多种途径之间的协同作用。例如,NK 细胞需要协同 IL-18 (NF-κB) 和 IL-12 (STAT4) ,才能最大程度地表达 IFN-γ。
3、STAT之间的协同和对抗
无论两个信号的异同,不同的 STAT 分子都可以相互协同或拮抗,从而影响转录。这些作用可以通过物理相互作用、AD 的丝氨酸磷酸化、协同 DNA 结合和特定信号抑制剂而产生,STAT1 和 STAT3 是其中典型的代表。由于 STAT1 和 STAT3 的丝氨酸磷酸化,IL-2 和 IL-12 介导的协同作用,导致更高水平的 IFN-γ 表达。这些 STAT 之间的拮抗作用可见于IFN 和 STAT3 激活细胞因子状态下,这一过程影响多种疾病的发生发展。例如,在自身免疫性多内分泌病-念珠菌病-外胚层营养不良 (APECED) 、STAT1 功能获得 (GOF) 及慢性皮肤黏膜念珠菌病 (CMC),均依赖于 STAT1 及STAT3 介导的免疫反应。在上述疾病中,STAT1 和 STAT3 被相同的细胞因子(即 IL-6、IL-21 和 IL-27)激活。但是,这些 TF 拮抗作用的潜在机制更加难以捉摸。目前,一些学者已经努力试图解决这个难题,在 IL-6 和 IL-27 存在的情况下,STAT3 负责转录输出,而 STAT1 是特异性的驱动因素。在 CD4 + T 细胞中,通过 IL-21 信号传导,STAT1 和 STAT3具有相反作用。
STAT1 和 STAT4 也可以相互拮抗。例如,IL-12 和 IL-18 可以通过降低 STAT1 转录水平,以及 I 型 IFN 转录输出,从而拮抗 IFN-α 信号传导。然而,在 NK 和 CD8 T 细胞中, I 型 IFN高表达的情况下,STAT1 和 STAT4均被激活。然而在低表达IFN-γ的NK 细胞和 CD8 T 细胞中, STAT1 和STAT4具有拮抗作用。在病毒感染其间,这种效应通过STAT 水平的改变而改变。与 STAT1 相比,STAT4 水平最初较高,但随着感染期间 STAT1 蛋白水平的增加,STAT1 拮抗功能开始发挥作用,可能是为了限制潜在的破坏性反应。
此外,经过IFN-I 诱导后,ISGF3(STAT1、STAT2、IRF9 复合物)和 γ 激活因子(GAF、STAT1 二聚体)之间会发生拮抗作用。当 IFN-I 信号通路因 STAT2 或 IRF9 缺陷而受损时,USP18 等负调节因子的诱导作用就会消失,从而导致 GAF 转录反应延长。STAT 的拮抗作用有助于解释细胞因子信号传导的特异性。然而,具体机制仍然需要深入研究。
细胞因子作用
生物学技术的进步,促进了细胞因子和 IFN 作用的新发现。通过微阵列技术,研究者全面定义了由细胞因子和 IFN 诱导的整个转录组,但是目前,大规模测序平台取代了微阵列技术。这些研究结果表明,细胞因子和干扰素不仅诱导了数千个基因,包括参与宿主防御反应的基因,而且还诱导了编码调节细胞周期和新陈代谢的基因。在此期间,通过使用染色质免疫沉淀测序 (ChIP-seq) ,可以全面定义整个基因组中的 STAT 结合位点,这项工作揭示了数以万计的细胞因子诱导的基因组结合位点。尽管很多研究工作都致力于枚举传统的蛋白质编码基因,但目前最新的研究结果证实,大部分基因组以细胞和激活特异性方式转录,并且 microRNA 和长链非编码 RNA (lncRNA) 的数量超过常规基因。据一些学者推测,STATs 对所有这些转录本都有重大影响。
1、STAT 调节的增强子
增强子是在其基因组、细胞和有机体环境中,与通过顺式调节机制转录蛋白质相互作用的DNA区域。增强子与 STAT ChIP-seq 一起,针对染色质修饰或 HAT p300 的抗体,与 STAT 缺陷型活化细胞一起用于研究STAT 的基因组影响。用于测量基因组的方法,可以评估少量细胞甚至单个细胞,从而绘制体内具有活性细胞,而无需在体外进行大量操作和扩增。这种技术进步,进一步加速了研究者对 STAT 基因组的理解。
很明显,STAT 与基因组的结合具有多重后果。转录组学“IFN 特征”相关的疾病(即干扰素病),与 IFN 表观基因组特征相关。IFN-γ 和 IL-4可以相互抑制表观基因组和转录组的改变。因此,STATs 也可以介导诱导表观基因组和转录组效应的改变。
活性增强剂可对STAT产生广泛影响(图 6)。它们可以影响组蛋白修饰,并且不会被谱系定义 TFs (LDTFs) 的作用所取代。除此之外,活性增强剂还可以参与潜在增强子的形成,这些增强子在细胞刺激后变得活跃。另一方面,生物信息学的发展,推动了缺乏 LDTF 结合基序的区域的可访问性,LDTFs在与 STATs 的关联区域中富集。对于未标记(潜在)增强子,LDTF 和 STAT 可访问性的先后顺序具有争议性。因此,一些学者这 LDTF 作为 STAT 的“先锋因素”提出了质疑。与 STATs 在建立增强子识别方面优于 LDTFs 一致,JAK 抑制可防止组蛋白标记和 PU.1 募集。因此,LDTF 和 STAT 在两个方向上的相互依赖性提供了组合变异性,有助于基因程序的特异性研究发展。

图6:STATs 和染色质组织。STATs在表观遗传标记的启动子和靶基因的增强子上与DNA结合,以调节转录。在启动子(H3K4三甲基化)、增强子(H3K27乙酰化)和RNA Pol II转录区(H3K36三甲基化)上观察到不同的组蛋白表观遗传标记。例如,Ifng位点,它由一个超级增强子调控,这些增强子被并富集于STATs、T-bet和p300,以响应细胞因子刺激。共聚焦显微镜(左上角)记录了STAT转录核凝析物的信号依赖性聚集。超级增强子、转录凝聚物和拓扑关联域(TADs)都可能汇聚成一个功能单元,建立一个物理平台来调控靶基因的动态转录。
控制细胞特异性功能的关键基因,受到由 TF、共激活因子、RNA Pol II 和延伸因子密集占据的增强子簇的调节。目前最火热的相关研究结构,是一种称为超级或拉伸增强剂 (SE) 的结构(图 6)。在 B 细胞中,对于细胞增殖、存活和分化,STAT5 与 SE 的其他 TF 网络之间的平衡是关键影响因素。总之,STATs 可以通过多种机制来发挥作用——直接转录诱导/抑制、表观遗传调节或兼而有之, STAT 对表观遗传的调节有很大作用。
STATs 的线粒体定位
STAT除了在细胞质和细胞核中具有突出作用外,一小部分 STAT1、STAT3 和 STAT5(占总数的 5%–10%)存在于线粒体中,其功能独立于细胞质信号传导和核基因激活。在线粒体中, STAT3 的研究成果最多,因为它通过调节呼吸、氧化还原稳态和线粒体转录直接促进 Ras 诱导的转化。在 Ras 转化的细胞中,ATP 水平降低,从而导致电子传递链的复合物 II 和 V 的活性降低。酪氨酸 705 磷酸化对 STAT3 的功能至关重要,它不需要驱动其线粒体功能,但似乎影响STAT3 在 S727 位点上的磷酸化。STAT可以进入线粒体以及其他细胞器,其在细胞器功能仍然需要深入研究。
JAKs 和 STATs 的体内功能
小鼠的基因研究为 JAK、STAT 和相关分子的作用提供了深刻的见解,相关基因的突变和变异揭示了与人类疾病的相关性(图 2)。在以下疾病中,可观察到JAK-STAT 通路突变(表 1)。
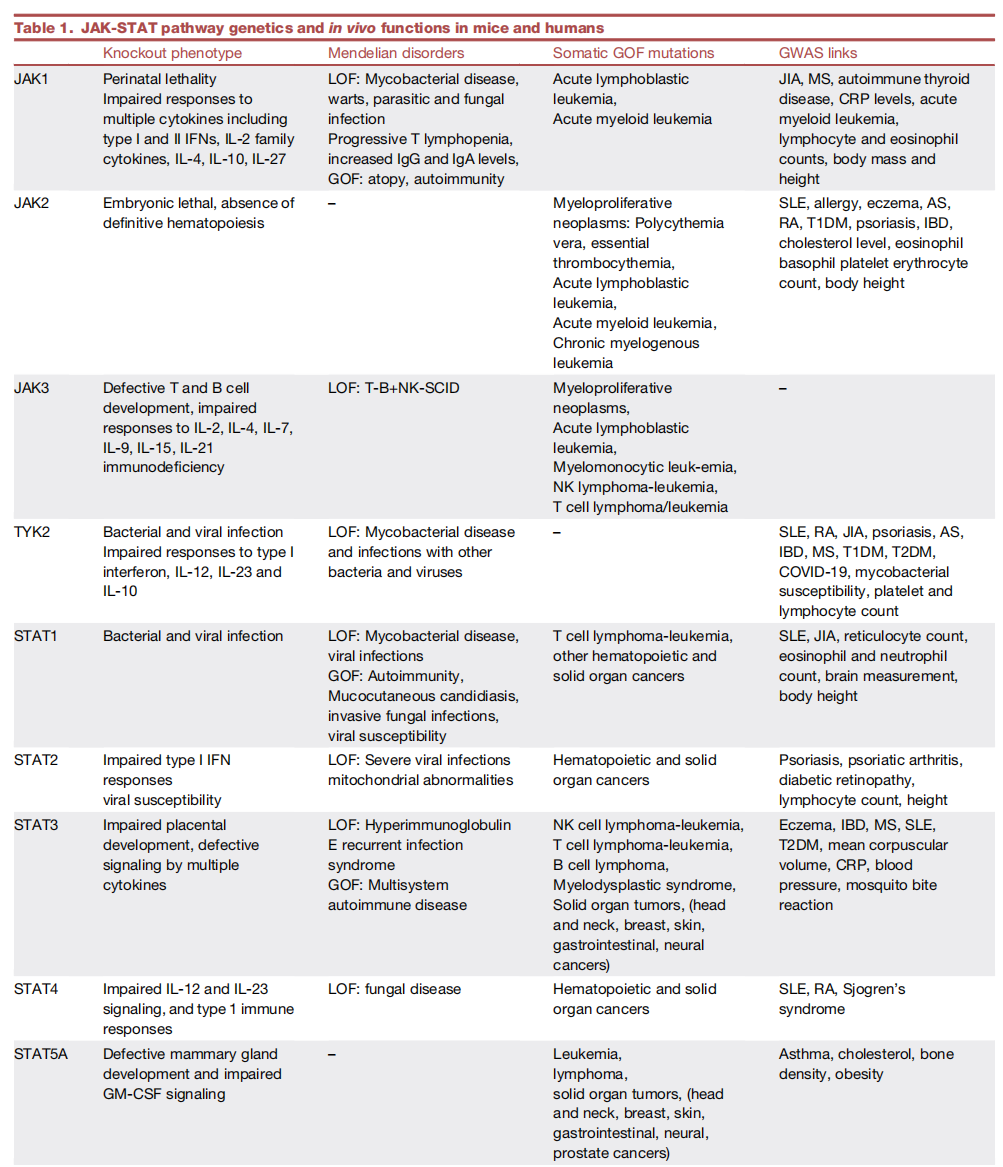

表1:JAK-STAT 通路在小鼠和人类疾病中的功能。AS,强直性脊柱关节炎;CRP,C反应蛋白;GOF,功能增益;IBD,炎症性肠病;JIA,幼年特发性关节炎;LOF,功能丧失;MS,多发性硬化症;RA,类风湿性关节炎;SCID,严重联合免疫缺陷;SLE,系统性红斑狼疮;T1DM,1 型糖尿病;T2DM,2型糖尿病。
1、JAK 突变
JAKs 的关键作用首先在联合免疫缺陷 (SCID)综合征中得到证实。在SCID中,编码 γc 的IL2RG突变是SCID 发生的分子改变。由于 JAK3 与 γc 相关联,已有研究证实,JAK3 常染色体隐性功能丧失(LOF)突变是导致 SCID 的重要原因。值得注意的是,具有LOF突变的患者和小鼠在其他器官系统方面都是正常的,这表明 JAK3 与其他 JAK 相比,具有非常特殊的功能。
在TYK2 缺陷小鼠中,有研究证实,细胞因子反应的抑制具有选择性,研究最为充分的是 IFN 和 IL-12/23 家族细胞因子反应的抑制。这些细胞因子反应抑制后,机体对感染的易感性增加。人类的 TYK2 缺乏与细菌和病毒感染以及过敏性疾病有关,与此同时,人类TYK2的 LOF 变体(P1104A) 与自身免疫性疾病发病率降低和结核病风险增加有关。
JAK2 对包括造血生长因子在内的许多细胞因子具有重要作用。在JAK2缺陷小鼠中,胚胎在子宫内多因骨髓衰竭而死亡。在一系列关于骨髓增生性肿瘤 (MPN)研究中,包括真性红细胞增多症、原发性骨髓纤维化和原发性血小板减少症,可发现JAK2 GOF 突变。最常见的突变是位于 JAK2 PK 结构域的 V617F位点,这导致细胞因子增殖。其他与 MPN 相关的突变位点包括 R683G、R683S 和 R683T。在果蝇肿瘤致死 (Tum-1) 突变中,首次发现JAK GOF 突变,该突变导致血细胞恶性瘤的形成。截至目前,全长 JAK 结构的明确,为PK 突变导致细胞因子非依赖性信号传导机制的研究提供了可行性方法。最近的一项研究发现,GOF 突变位于 PK 中间二聚体界面的核心,可以增强包装互补性,从而促进配体非依赖性激活。
与依赖PK的许多细胞因子一致,JAK1 缺陷小鼠在围产期内死亡率升高,同时,多器官发生损伤。在人类中,JAK1 LOF 突变与复发性分枝杆菌感染、发育迟缓和早发性转移性膀胱癌相关。与此同时,JAK1 GOF 突变导致自身炎症相关疾病和超免疫球蛋白 E 综合征 (HIES)。随着全基因组关联研究 (GWAS) 的发展,研究中揭示了JAK1在幼年特发性关节炎 (JIA)、多发性硬化症 (MS)、自身免疫性甲状腺疾病、C 反应蛋白 (CRP) 水平、淋巴细胞和嗜酸性粒细胞计数中的多态性。
2、STAT 突变
尽管 STAT1 被许多细胞因子激活,但 STAT1 敲除 (KO) 小鼠对细胞内病原体(细菌和病毒)的易感性增加。此外,STAT1 对造血前体功能很重要。STAT1-GOF突变患者易患自身免疫性疾病,并且对真菌和病毒的易感性增加;然而,STAT-GOF 突变的分子作用机制仍然不是很清楚。有学者猜测,GOF 突变可能有利于 STAT1 的平行磷酸化状态;然而,这一猜测尚未通过详细的结构分析得到证实。
小鼠和人类的STAT2 LOF 突变,使得机体对病毒的易感性增加。然而,一些 STAT2 位点的突变不影响磷酸化或活化;相反,这些位点突变后,会对 STAT2 将 USP18 募集到 IFN 受体亚基 IFNAR2这一过程产生抑制作用。由于 I 型 IFN 信号的负调控缺陷,有学者指出,这些 STAT2 突变可能与严重的干扰素病有关。
STAT3 的种系缺失在小鼠中是致命的,因为它在胎盘中具有重要功能。相关基因敲除小鼠模型的发展,揭示了 STAT3 在多种组织中的广泛作用。相关大量研究证实,STAT3在先天性和适应性免疫、干细胞、伤口愈合、组织修复和癌症中具有重要作用。STAT3的LOF突变是高免疫球蛋白 E(乔布氏)综合征的分子基础,这是一种原发性免疫缺陷综合征,与颅面、骨骼和血管异常有关。STAT3 GWAS 变异与炎症性肠病 (IBD)、MS、2 型糖尿病 (T2DM) 和心肌梗塞有关,具有STAT3 GOF 突变的患者好发淋巴造血系统肿瘤和多发性自身免疫性疾病。
STAT4敲除小鼠的 IFN-γ 产生受损,并且对弓形虫的易感性增加。在人类中,STAT4 LOF 突变与真菌感染和病毒的易感性有关。目前一些研究也发现,STAT4变异与狼疮、类风湿性关节炎 (RA) 和干燥综合征相关。此外,STAT4编码变体(Glu128Val)的突变使 RA 的风险加倍。
在基因组中,相邻的 STAT5A 和 STAT5B 具有超过90% 的相同性。Stat5a 敲除 小鼠缺乏催乳素介导的乳腺发育,而Stat5b 缺陷小鼠的性腺发育受限。Stat5a/b双缺陷小鼠大多数在出生后几周内死亡,出生的小鼠具有不育,乳腺发育缺陷,骨髓细胞不足。人类的STAT5B LOF 突变不仅导致生长障碍、面部畸形和自身免疫,而且还可以可导致非克隆性嗜酸性粒细胞增多症、湿疹、荨麻疹和腹泻。STAT5A突变与肥胖有关,而目前关于STAT5BGWAS相关研究,揭示了与淋巴细胞和中性粒细胞计数、哮喘和过敏以及骨密度的联系。
与 STAT6 在 IL-4 和 IL-13 信号传导中的作用一致,缺乏 STAT6 的小鼠无法产生 2 型免疫反应,从而导致寄生虫消除受损。一项研究发现,STAT6 GOF 突变与伴有嗜酸性粒细胞增多症的过敏性疾病相关,包括过敏、哮喘和嗜酸性粒细胞增多症。
3、负反馈调节器的突变
在小鼠中, SOCS1 的缺失对 IFN-γ 过敏是致命的,而在人类中, SOCS1 缺陷表现出早发性自身免疫和过敏性疾病。人类缺乏 USP18 和 ISG15 ,易患严重的全身性干扰素病。
JAKs 和 STATs 在癌症中的复杂作用
在发现该通路后不久,就有许多研究论证了JAK-STAT通路在癌症中的作用,特别是病毒介导的肿瘤。在这一过程中,研究者发现了许多与癌症相关的 JAK 和 STAT。然而,在癌症中, JAK1突变失活可以消除 IFN-γ 信号传导,逃避免疫细胞杀伤,并改变肿瘤微环境中的免疫细胞功能。细胞因子和 JAK-STAT 通路在癌症和肿瘤微环境中的作用较为复杂,这方面的研究具有挑战性。
在肿瘤中,最早研究的是GOF 突变。一些早期研究发现, Tel-JAK2 融合蛋白和其它JAK2 融合蛋白(例如,PCM1、SEC1A 和 PAX5)与白血病相关。此外, STAT3的SH2 结构域发生二聚化、与 DNA 结合,激活转录,从而导致血液细胞的癌变。在造血系统恶性肿瘤中, STAT5 的 SH2 结构域突变也较为常见。最近的一项研究证实,STAT5 的N642H结构域突变,可以驱动小鼠白血病的发生。
随着测序技术的进步,人们对 JAK/STAT 相关分子的突变有了更全面的了解。JAK 和 STAT 的突变很常见,但主要见于造血/淋巴恶性肿瘤。例如,STAT1 是白血病发展中的肿瘤启动子。在 B 细胞急性淋巴细胞白血病 (ALL) 中,常见 STAT5 和细胞外信号相关激酶 (ERK) 突变,然而,这两种突变途径具有拮抗功能。STAT5 通过对靶基因的反向调节,拮抗 NF-κB 和 IKAROS的功能,这一过程与TF 网络分子相关,包括 PAX5、EBF1、PU.1、IRF4 和 IKAROS。此外,STAT5B的N642H位点突变是一种频繁发生的驱动突变,可促进侵袭性 T 细胞白血病/淋巴瘤的发生。此外, 在T 细胞白血病中,STAT5 与 NF-κB 或 IKAROS 比例高的患者具有不良预后,但STAT5不同位点的缺失也加速了白血病的发生——这方面仍然需要深入研究。
在非造血肿瘤中(乳腺癌,肺癌,结直肠癌),虽然 JAKs 和 STATs 的突变发生不是很常见,但是,这并不意味着该通路与实体瘤无关。除了位点突变之外,STAT3 和 STAT5 的激活在许多人类癌症中较为常见,并且 STATs 的过表达与不良预后相关。STAT3 和 STAT5,可以直接调节促进增殖的基因表达(细胞周期基因,如 MYC),抑制细胞凋亡相关基因表达(BCL2、BCLXL、MCL1 和 BIRC5)。此外,STAT3 和 STAT5 还可以通过经典和非经典机制,促进肿瘤干细胞增殖、远处转移和改变新陈代谢。除了激活酪氨酸磷酸化之外,STAT3 的功能也可以通过丝氨酸磷酸化来介导。在肺腺癌模型中,Ras 依赖性 STAT3 丝氨酸磷酸化是重要的肿瘤驱动因素。
除了细胞内在作用(即 JAK 和 STAT 在肿瘤细胞本身中的作用)外,STAT3在炎症、伤口修复和纤维化中也发挥调节作用。在肿瘤微环境和基质癌相关成纤维细胞中,STAT可以通过改变细胞外基质的产生、血管生成和代谢环境来驱动肿瘤的发展和进展。

影响癌细胞生长和存活的I型干扰素(IFN-I)依赖性途径。图片源自于Cheon H J, Wang Y, Wightman S M, et al. How cancer cells make and respond to interferon-I[J]. Trends in Cancer, 2022.doi: 10.1016/j.trecan.2022.09.003.
干扰素在癌症中具有复杂的作用。2022年,发表于《Trends in Cancer》杂志上的一篇综述总结了癌细胞如何控制自身合成 IFN-I ,以及这些细胞如何控制对存在于肿瘤微环境中的 IFN-I 的反应。此外,最近一些研究,对 IFN-I 作为癌症免疫系统调节剂的重要性进行了很好的论证。在癌细胞中,高浓度 IFN-I 的促凋亡和抗生长活性减弱,低浓度IFN-I反而可以促进肿瘤的生长。在许多肿瘤中,缺乏酪氨酸磷酸化 (U-STATs) 和 IRF9 的 STAT1 和 STAT2 高水平表达,形成 U-ISGF3,驱动ISG 的表达,编码重要的促肿瘤蛋白。U-ISGF3 水平在正常细胞中较低, IFN-I可促进U-ISGF3的升高。在许多癌症中,高水平的 U-ISGF3 通过诱导“IFN 相关 DNA 损伤抗性基因“的表达,或 IRDS 的表达,从而抑制药物导致的DNA 损伤。在肺癌细胞中,高水平U-STAT2 促进肿瘤细胞对顺铂的耐受性。
PD-L1 通常在肿瘤细胞表面表达,并因其在灭活细胞毒性 T 细胞中的重要性而广为人知,它还具有重要的肿瘤细胞内在活性。在肿瘤细胞内,PD-L1可以抑制具有细胞毒性的高水平 IFN-I反应,并且维持有益于肿瘤生长的低水平 IFN-I反应。细胞因子和 JAK/STAT 通路在癌症中的作用具有复杂性。
JAK/STAT 通路的靶向治疗
1、JAK抑制剂
目前大量的研究已经证实,JAKs 在一些疾病中具有重要作用。这引发了人们将 JAKs 作为治疗靶点的考虑。十种JAK抑制剂现在被批准用于治疗自身免疫性疾病和一些肿瘤性疾病(表 2)。

表2:批准的JAK抑制剂
Tofacitinib 是第一个被批准使用的JAK抑制剂,主要应用于器官移植后的同种异体移植排斥反应。基于 JAK2 GOF 突变的发现,JAK1/JAK2 抑制剂鲁索替尼第一个获得批准,用于治疗 MPN。随后,Fedratinib 和 pacritinib 也被批准用于 MPN。2021年,鲁索替尼也被批准用于急性和慢性移植物抗宿主病,外用鲁索替尼被批准用于特应性皮炎和白癜风。Tofacitinib 是第一个被批准用于RA和 IBD。随后,Baricitinib 被批准用于治疗 RA、斑秃和 COVID-19感染。其他几种 JAK抑制剂现在已被批准用于其他形式的免疫介导的关节炎、过敏性疾病和 IBD。奥拉替尼被批准用于治疗系统性红斑狼疮 (SLE)及干燥综合征。
迄今为止,JAK抑制剂治疗中观察到的大多数不良事件可以用特定细胞因子的作用机制来解释,并且具有较高的安全性。报道的不良相互作用包括血栓栓塞事件风险增加、血脂改变和心血管并发症。
开发选择性 JAK 抑制剂的动机是干扰较少细胞因子的功能,增加特异性,并且具有较少副作用。这些药物包括 JAK1 抑制剂(upadacitinib、filgotinib 和 abrocitinib)、TYK2 抑制剂(ropsacitinib/PF-06826647 和 NDI-031407和 deucravacitinib,一种针对 PK 结构域的变构 JAK2 抑制剂。目前,Deucravacitinib 被批准用于治疗斑块状银屑病。药物研究人员于2021年合成了一种靶向 PK 结构域的变构 JAK1 抑制剂。此外,药物研究人员正在努力合成局部应用及可吸入的JAK选择性抑制剂,以减少与全身给药相关的副作用。目前,一些药物专家指出,靶向蛋白质降解现在也是合成 JAK1 在内的激酶抑制剂的一种选择。
2、STAT 抑制剂
已采用各种策略来开发 STAT 抑制剂,包括干扰 SH2 介导的二聚化、DNA 结合或反式激活和诱导 STAT 降解。多种抑制剂正在临床试验中,但目前还没有药物被批准。
3、SOCS 模拟物
SOCS 肽模拟物已在免疫和肿瘤临床前模型中显示出疗效。
未来的挑战和机遇
30 年前,JAK-STAT 通路的发现,给人类带来了大量影响健康的信息,包括 COVID-19 的治疗。然而,不明确未来的挑战会对将来的研究产生误导。在下面的简短部分中,作者思考了几个主题,这是目前关于该通路研究的局限性。
1、结构和细胞生物学 JAK、STAT 和细胞因子信号
与 JAK1 和 STATs 有关的结构发现具有启发性,但细胞因子如何与 STATs 结合,这方面的研究仍是空白。随着成像方式的进步,我们可以使用更复杂的工具来分析细胞因子信号传导的早期事件,包括从膜到细胞核的 STAT 激活和转录及调控之后。STAT 磷酸化诱导二聚化激活核易位,这是STAT作用的典型模型,然而这一模型没有考虑到在细胞因子的状态,从而导致模型在一些方面具有争议性。STAT 蛋白与多种细胞器的关联尚未在功能和物理上得到充分认识。
2、更全面地了解 STAT 介导的染色质修饰和转录调控
增强子结构、增强子转录、lncRNA、组蛋白修饰、染色质环化和拓扑关联域 (TAD) 中的相互作用为基因表达的精确组织特异性、时间和动态调节提供了许多机会(图 6)。随着新型分子技术的发展,细胞因子和 STATs 对这些事件的影响正变得越来越清晰。尽管 STATs 是信号依赖性 TFs,但非磷酸化 STAT1 的作用,超出了作为信号依赖性 TF 的作用。关于U-STAT1 如何修饰染色质和三维基因组的研究仍是空白,需要深入探索。
解释增强子在染色质环之外的功能,以及增强子和启动子接近的技术模型是液-液相分离 (LLPS)。当TF 在确定的调节区域内,辅激活因子募集和具有内在无序区域的转录因子形成复合物,从而引发分层和凝聚物的形成。STAT 似乎参与了该过程(图 6),并具有分层特性。利用体细胞 GOF STAT 突变,详细了解 STAT 对相分离的生物物理方面的结构和功能贡献将很重要。截至目前,核 JAK2 的作用及其磷酸化组蛋白这一研究领域是空白。
3、实现细胞、组织和刺激特定程序
目前,最大的挑战是了解细胞因子产生的信号如何在JAK -STAT 通路中被放大的具体机制。目前这一领域的研究具有挑战性,破译细胞信号如何提供特定指令并不是细胞因子独有的问题,相反, JAK-STAT 途径可能具有许多可以解决这一基本生物学难题的属性。
首先要考虑的是信号成分表达的特异性和调控特点。JAKs 和 STATs 广泛表达,但在一些条件下,具有更高的选择性,例如 JAK3、TYK2 和 STAT4 优先在免疫细胞中表达。STATs 也直接、动态地调节它们自己的表达,STAT1 就是一个特别好的例子。在感染过程中,STAT4 最初介导 I 型 IFN 信号,但后来 STAT1 占主导地位。除了细胞因子受体,还需要考虑 JAK、STAT、SOCS 蛋白和其他下游元素的动态表达。在这方面,信号的强度可以在发育过程中或在感染过程中急剧变化。
其次,目前已经研究STAT 激活动力学,但大多数细胞因子激活不止一个 STAT 家族成员,且不同的STAT具有不同的激活幅度、磷酸化和去磷酸化速率,这些措施往往没有得到全面和严格的确定,因此需要改进技术来定量评估这些变化。值得注意的是,已经产生了可以改变这些变量的工程细胞因子和细胞因子拮抗剂,并可能成为有用的工具。STAT 异二聚体的形成被认为是解释特异性的另一种方式,但经常被忽视。尽管对 STAT 结合的基序的基因组分析总是揭示典型的回文基序,但相当大比例的 STAT 结合区域偏离了经典基序。在没有同源 DNA 结合基序的情况下如何招募 STATs 仍有待阐明。
第三,如前所述,可能会出现许多刺激特异性 STAT 翻译后修饰状态,这些状态会影响细胞内运输、DNA 结合和转录潜力;然而,这些修改通常不是全面和动态测量的。截至目前,几乎没有提供关于 JAK 响应不同刺激的翻译后修饰的相关研究。这是一个需要进一步研究的重要课题。
最后,细胞因子受体的多样性允许它们参与其它信号通路(例如,MAPKs、PI3K 等),这有相当大的可变性。在发育、炎症或其他事件中,细胞不受单个细胞因子的影响——多种外源性、内源性和诱导信号参与了调节过程。在这方面,STATs 与其他通路及拮抗通路如何协调,仍需深入研究。除此之外,还有增强剂的复杂性和多种因子密集结合的研究仍需进一步深入。
尽管需要复杂的、特定于上下游调节来实现特异性,但实现信号的特定解释的明显解决方案涉及组合逻辑。尽管这似乎是一个具有挑战性的研究内容,但多组学的应用,有助于全面测量通路随时间的变化量,这需要数量较少的单细胞就可完成。基于先例,对 JAK 和 STAT 的更复杂知识无疑将为具有更高特异性的新药提供许多机会。
原始出处:
Philips RL, Wang Y, Cheon H, Kanno Y, Gadina M, Sartorelli V, Horvath CM, Darnell JE Jr, Stark GR, O'Shea JJ.The JAK-STAT pathway at 30: Much learned, much more to do.Cell . 2022 Oct 13;185(21):3857-3876. doi: 10.1016/j.cell.2022.09.023

