“第四掌骨征”代表什么?
时间:2022-12-22 20:59:10 热度:37.1℃ 作者:网络
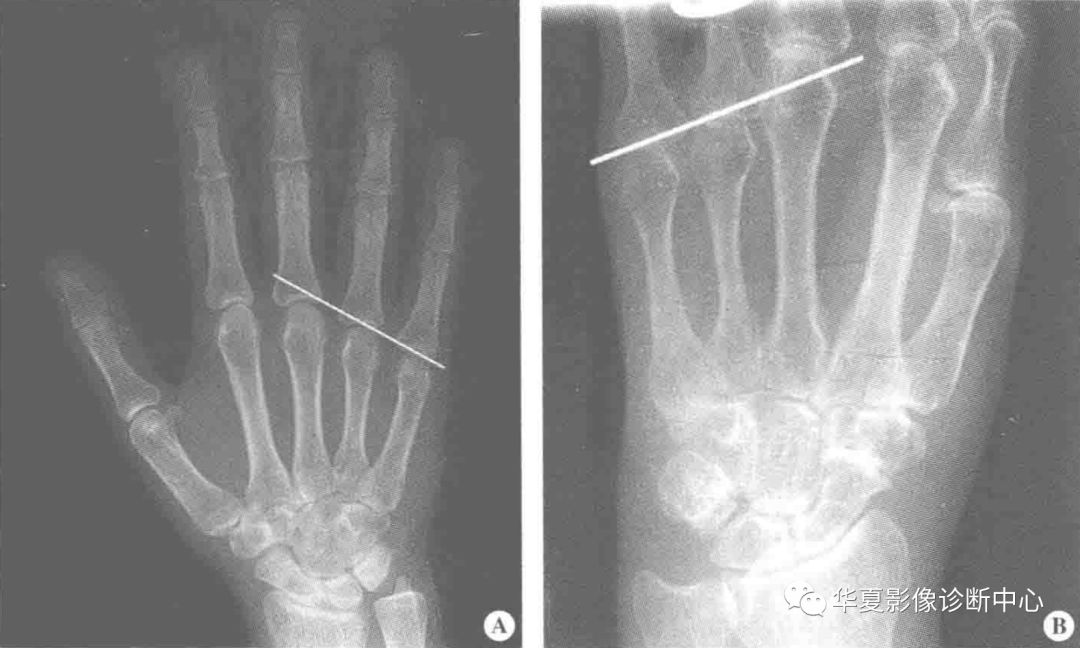
正常手的X线正位第4、5掌骨顶端的连线不与第3掌骨头相交,而在其远侧横过,如此线横贯第3掌骨头则为掌骨征阳性。
病理基础
第四掌骨征主要见于患有假性甲状旁腺功能减退症的患者中,也可在特纳综合征、遗传性多发性外生骨疣、同型胱氨酸尿症及其他少见类型的综合征中见到。需要和陈旧性骨折后改变、其他的手部创伤或外科手术后的表现相鉴别。
 疾病介绍
疾病介绍
假性甲状旁腺功能减退症(pseudo-hypoparathyroidism,PHP)是一种具有甲状旁腺功能减退症的症状和体征的遗传性疾病,其遗传方式主要为X染色体显性遗传,近年来发现基因缺陷也可在常染色体上,为显性或隐性遗传。本症主要是靶器官(骨和肾)对甲状旁腺激素失敏,甲状旁腺增生,血中甲状旁腺素增加,而临床表现为甲状旁腺功能减退,典型病例还有独特的骨骼和发育缺陷。
临床分类及表现
根据终末靶器官对PTH抵抗发生在 cAMP生成之前或之后,PHP可分为I型和II型,而PHP-I型又根据存在或缺乏Albright遗传性骨营养不良 (Albrighthereditaryosteodystrophy,AHO)(其主要特征为圆脸、矮胖、指/趾骨畸形、异位钙化及/或智力障碍)、Gsα的活性是否正常等又可进一步分成Ia、Ib和Ic3个亚型。
(1) PHP-I型
①PHP-Ia型:是临床最常见的类型,也称为Albright遗传性骨营养不良症(AHO),典型体征包括身材矮小、肥胖、圆脸、短颈、盾状胸、短指(趾)畸形等,掌骨X线检查可见第4与第5掌(趾)骨较短。AHO患者血细胞Gsα活性降低,存在多发性内分泌缺陷,如性腺功能减退、TSH抵抗、生长激素缺乏等,部分患者伴有智力低下。
靶细胞膜受体-腺苷酸环化酶系统缺陷,对PTH不反应,受体不能与PTH结合或者能够结合但不产生cAMP,cAMP的缺少使PTH不能发挥激素的生理效应。尿cAMP降低甚至测不出,注射活性PTH后其尿cAMP和尿磷不增加。
②PHP-Ib型:患者无AHO畸形,以肾脏对PTH抵抗为主,Gsα活性正常。这种类型可能是PTH受体的缺陷所致,但产生PTH受体活性下降的分子学机制还不明确。
③PHP-Ic型:临床极少见,有AHO畸形并伴有多发性激素抵抗,其发病机制尚不明确。
(2) PHP-II型
患者无AHO畸形,仅表现为PTH抵抗,尿cAMP升高或正常,注射外源性PTH后,尿cAMP进一步升高,但尿磷排出量无明显变化。
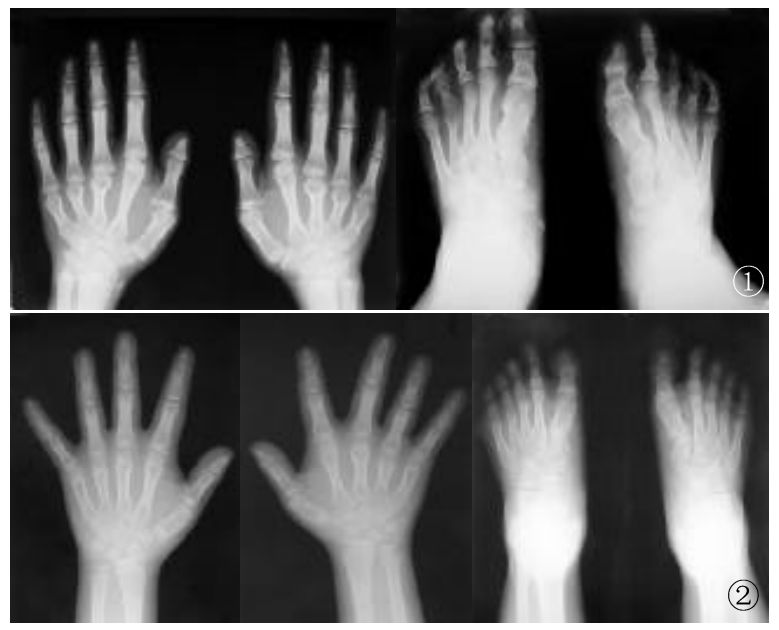
图1 X线片示第三、五短指(趾)畸形
图2 X线片示第四、五短畸形,无短趾畸形
发病机制
目前研究提示与GNAS1基因缺陷相关,该致病基因定位于20q13.11,其主要与编码鸟核苷酸结合蛋白(GTP结合蛋白)的α亚基(Gsα)有关,涉及GNAS1基因的基因组亲缘印迹。正常情况下,PTH与靶细胞膜上特异性受体结合后,从而诱导受体构象改变,激活G蛋白,使得与α亚单位结合的GDP释放,此时GTP再与α亚单位结合,又使得形成的α-GTP与βγ二聚体分离,激活腺苷酸环化酶(AC),生成第二信使cAMP,进一步激活蛋白激酶(PKA),继而催化蛋白质与酶的磷酸化,促进肾小管对钙的重吸收,使尿钙减少,血钙升高。而在PHP患者中,由于GNAS基因的突变,使得Gsα在靶组织中的表达量下降或活性降低,从而干扰了第二信使cAMP的形成,则显示出对PTH的抵抗作用,进而使得甲状旁腺代偿性增生或肥大,PTH合成与分泌增加。研究证实大多数PHP患者存在G蛋白异常,仅有部分患者Gsα活性正常。
疾病检查诊断
▉实验室检查
1.尿液检查 尿钙及尿磷减少。
2.血生化检查 本病的血生化表现与真性甲状旁腺功能减退症相近似。
(1)血钙低、血磷高。
(2)血中甲状旁腺素(PTH)正常或增高,注射PTH 200U后尿中cAMP及磷不增加。可证明肾小管对PTH作用有抵抗性。
(3)血碱性磷酸酶正常。
▉其他辅助检查
X线骨骼检查可发现骨骼线融合早和颅顶骨增厚。
▉鉴别检查
1.特发性甲状旁腺功能减退 搐搦较重,异位钙化少见,无异常体形及短指(趾)畸形,血中PTH减低,不同的是该病注射PTH治疗有效。即尿中cAMP明显增加,尿磷排泄量可较注射前增加5~6倍以上,而假性甲状旁腺功能减退症者增加仅2倍。
2.假-假性甲状旁腺功能减退 本症实际上是假性甲状旁腺功能减退症的不完全型,可能是由于近端肾小管上皮细胞内cAMP依赖蛋白激酶异常或功能障碍所致,也可能与遗传有关。多在成年期出现症状,临床表现与假性甲状旁腺功能减退相似,亦有体态异常,但无抽搐。实验室检查,血钙和磷均正常,血清PTH正常,注射PTH试验反应正常。这些实验室检查结果,均与假性甲状旁腺功能减退症不同。
案例分享
患者,男,血液送至北京康旭医学检验所,检测项目:骨代谢检测包+MLPA065:GNAS基因大片段检测(包含甲基化)
临床表型:患者因反复肢体抽搐伴意识丧失2年入院,外院查 PTH 升高,血钙偏低。
临床关注或基因:假性甲状旁腺功能减退症,GNAS基因
检测结果:
1.未发现受检者GNAS基因存在大片段变异,发现相关甲基化异常;
2.未发现受检者其父母、妹妹GNAS基因存在大片段变异,也未发现相关甲基化异常。
结果说明:
1.GNAS是一个复杂的印记基因,定位于20q13.3。主要由编码刺激性G蛋白α亚单位( stimulatory G-protein alpha-subunit,Gsα) 的1~13外显子及其上游的启动子序列(如 Nesp,Nespas,GNASxl,A/B)组成。大量研究发现编码Gsα基因的外显子的杂合子失活突变可导致Gsα表达量下降或活性降低,从而引起激素抵抗作用的发生或Albright遗传性骨营养不良(AHO)的形成。另外,分子遗传学研究也发现有一部分PHP患者并不存在编码Gsα基因的外显子的突变,而表现为GNAS基因印记的缺陷,如外显子A/B差异甲基化区域( differentiallymethylated region,DMR)的甲基化缺失。其突变类型包括错义突变、无义突变、碱基对的插入或缺失(移码突变)。
2.GNAS基因既往报道与McCune-Albright综合征(McCune-Albright syndrome)相关,遗传方式尚未明确;也是假性甲状旁腺功能减退症Ia型(PHP-Ia)、假性甲状旁腺功能减退症Ib型(PHP-Ia)、假性甲状旁腺功能减退症Ic型 (PHP-Ic)、假性甲状旁腺功能减退症(PHP)的致病基因,均为常染色体显性遗传方式(AD)。
MLPA检测结果:
受检者:

受检者之父:

受检者之母:

受检者之妹:
 治疗
治疗
本症尚无有效的预防和根治方法,但如诊治及时,一般预后良好。急性发作期要迅速终止搐搦和痉挛,防止喉头痉挛导致呼吸困难甚至窒息死亡。非急性期治疗以防止急性发作和延缓病情进展为目的。口服钙剂及维生素D是有效的方法。血钙应控制在正常低值(2.0mmol/L~2.2mmol/L);近年来多使用活性维生素D制剂,效果更佳。伴有低镁血症的患者应及时补充镁剂,可以增加PTH对靶器官的敏感性,改善低钙症状。

