台湾省77例大疱性表皮松解症患者的基因突变分析
时间:2023-07-30 11:36:40 热度:37.1℃ 作者:网络
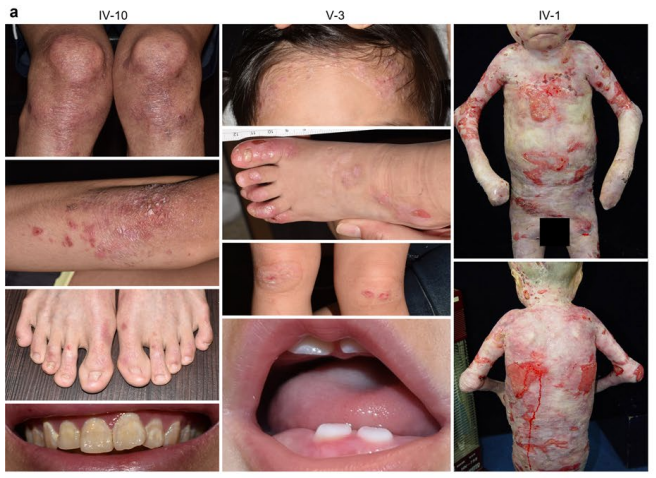
遗传性大疱性表皮松解症(Epidermolysis bullosa,EB)是一组罕见的异质性遗传病,是主要由于皮肤结构蛋白缺陷而导致的一组疾病,患上此病的人皮肤脆性增加,轻微碰撞、摩擦,皮肤上就会出现水疱、糜烂,继发感染,且容易引发严重并发症,如食道粘膜狭窄、手指并指挛缩、角膜瘢痕化导致眼睛失明等。
经典形式的EB是由于编码负责维持皮肤和/或粘膜细胞完整性和粘附蛋白质的16个基因中的一个发生突变,而另外24个基因的突变可能导致EB所包含的其他非经典疾病的皮肤脆弱。
除了皮肤粘膜脆性和瘢痕形成外,EB还会引起许多其他表现,包括胃肠道和尿道狭窄、贫血、发育不良、肌肉萎缩和皮肤恶性肿瘤。
随着透射电子显微镜(transmission electron microscopy,TEM)、免疫荧光显微镜(immunofluorescence microscopy,IF)研究和突变分析的引入,EB的研究和诊断在过去的50年里,我们对这种疾病的了解大大增加。
经典形式的EB分为单纯型EB(EBS)、交界型EB(JEB)、营养不良型EB(DEB)或Kindler综合征。
根据2020年EB共识重新分类,基于临床、病理和遗传发现患者可以进一步分为35种亚型。既往关于EB的临床表型和分子病理学的报道主要集中在欧洲、美国和中东人群。在亚洲,相关研究报道较少。
台湾省研究团队通过收集2012年1月至2021年6月国立成功大学附属医院EB患者的临床资料。诊断手段包括透射电子显微镜、免疫荧光研究和全外显子组测序(WES)。新的剪接位点突变的致病性通过皮肤mRNA的逆转录酶PCR测定,然后进行Sanger和/或RNA测序。
研究共纳入45个家庭的77例EB患者,单纯型EB 19例,交界型EB 6例,营养型EB 52例。38个家族中有37个(97.4%)鉴定出致病变异,其中WES被用作突变分析的一线工具,RNA测序确定了其余家族的致病变异。共鉴定出60个EB相关基因突变,包括22个新突变。这些突变涉及KRT5、KRT14、PLEC、COL17A1、LAMB3、LAMA3、ITGB4和COL7A1。
通过台湾省EB患者不同的临床表现和分子病理,扩大了我们对这种疾病的认识。WES是鉴别EB相关变异的有效一线诊断工具。当发现多个潜在致病性剪接位点突变时,RNA测序补充了WES进行诊断。
参考文献:
Tu Wei-Ting,Hou Ping-Chen,Chen Peng-Chieh et al. Mutational analysis of epidermolysis bullosa in Taiwan by whole-exome sequencing complemented by RNA sequencing: a series of 77 patients.[J] .Orphanet J Rare Dis, 2022, 17: 451.

