JACC-BTS 复旦大学附属中山医院邹云增/吴剑团队揭示NLRP3-TAK1介导焦亡与肥大反应的互作失衡触发压力超负荷心脏重构
时间:2023-08-11 17:32:59 热度:37.1℃ 作者:网络
心力衰竭仍然是全球具有高发病率和高死亡率的主要心血管疾病。心肌肥厚不仅是对心脏不良刺激的适应性反应,也是心力衰竭的独立危险因素。压力超负荷被认为是心脏代偿性肥厚反应的主要诱因。近年来炎症反应在压力超负荷心脏重构等非梗死性心脏病中日益受到关注,然而与炎症密切相关的细胞死亡如何介导心脏肥大仍有待阐明。
NLRP3(编码NOD-、LRR-和pyrin结构域蛋白3)是NLRP3炎症小体(由ASC、Caspase-1和NLRP3组成)的传感器蛋白。NLRP3炎症小体的形成导致依赖Caspase-1的促炎细胞因子IL-1β和IL-18的释放,以及GSDMD介导的细胞焦亡。
尽管大量研究表明,抑制NLRP3对心肌梗死或缺血再灌注引起的心肌损伤有显著的保护作用,但抑制NLRP3对压力超负荷引起的心肌肥厚的疗效却存在争议,有观点认为敲除NLRP3会加速肥大心肌从代偿转向失代偿。
转化生长因子β活化激酶1(TAK1)对于调控重要的生理过程十分关键,包括免疫细胞激活、炎症和心肌肥厚反应。TAK1是先天免疫和促炎信号的胞内中枢分子,其对心肌肥厚的影响仍有争议。既往大多认为TAK1是促进病理性心肌肥厚的关键因子,但近年来,也有发现敲除TAK1会产生明显的细胞毒性。因而,在炎症反应介导的环境下,NLRP3是否与TAK1互作精细调控压力超负荷心脏重构尚待阐明。
2023年8月9日,复旦大学附属中山医院邹云增/吴剑课题组在JACC Basic to Translational Science在线发表题为“TAK1 Activation by NLRP3 Deficiency Confers Cardioprotection Against Pressure Overload-Induced Cardiomyocyte Pyroptosis and Hypertrophy”的研究论文,揭示了NLRP3-TAK1信号轴对焦亡(炎性坏死)和肥大反应的双重调节作用,其互作失调触发压力超负荷心脏重构。

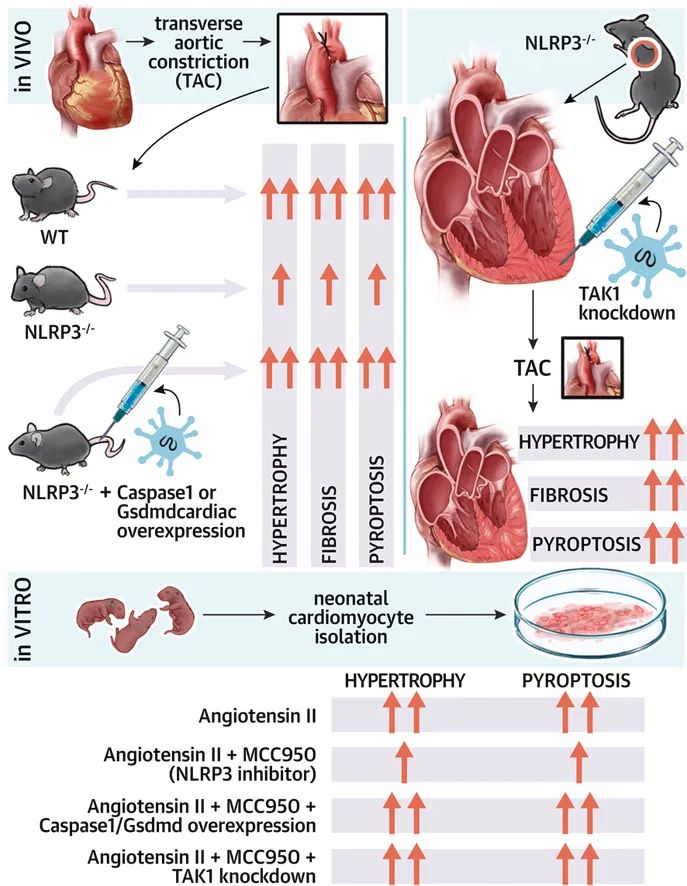
首先,研究者对NLRP3敲除小鼠行主动脉缩窄术(TAC)构建小鼠压力超负荷心肌肥厚模型,发现敲除NLRP3会减轻压力超负荷诱导的心肌焦亡,保护心功能。同时体外实验结果表明,MCC950 (NLRP3抑制剂)处理的小鼠乳鼠心室肌细胞,减轻了血管紧张素II(Ang II)诱导的心肌细胞焦亡与肥大。
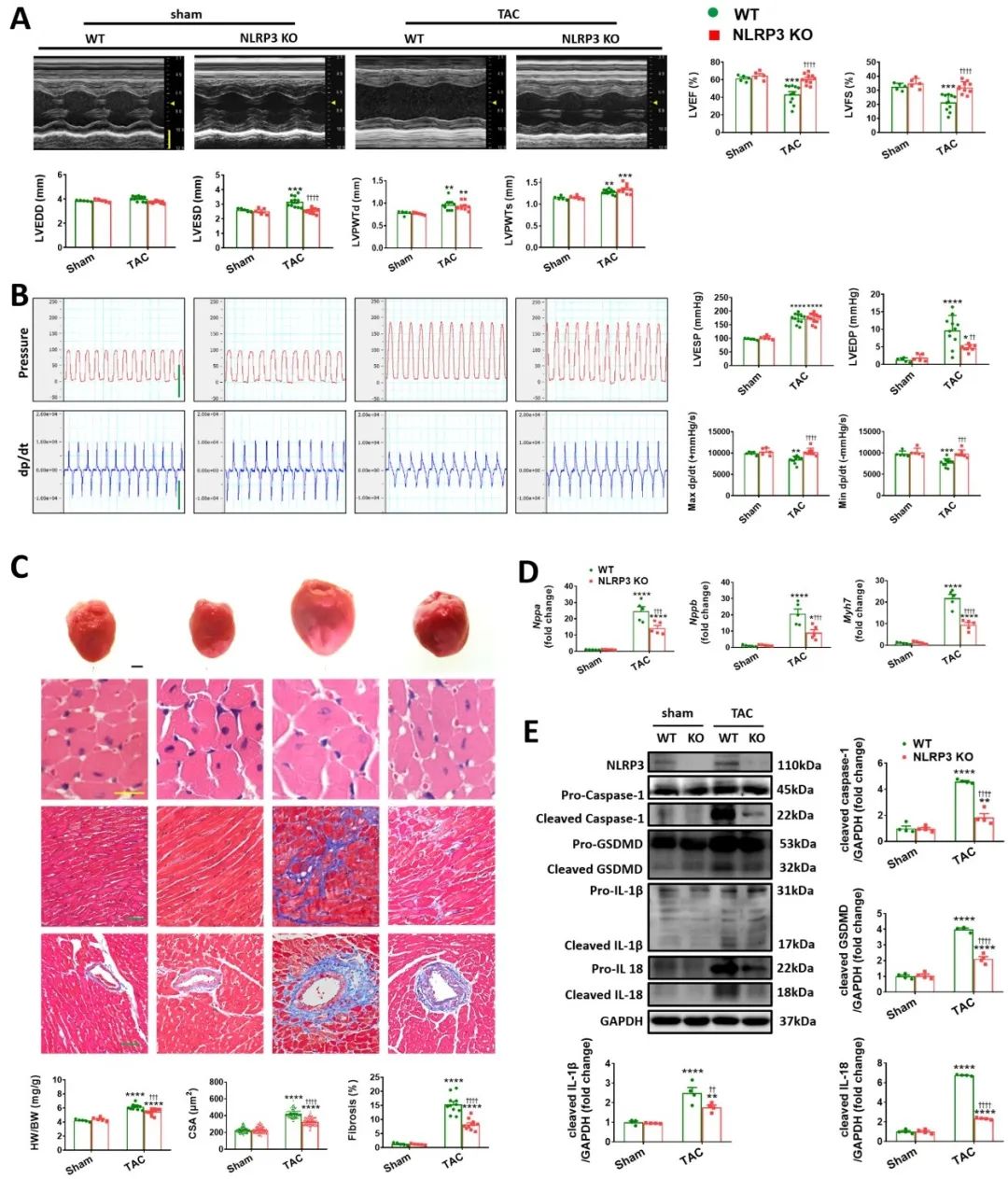
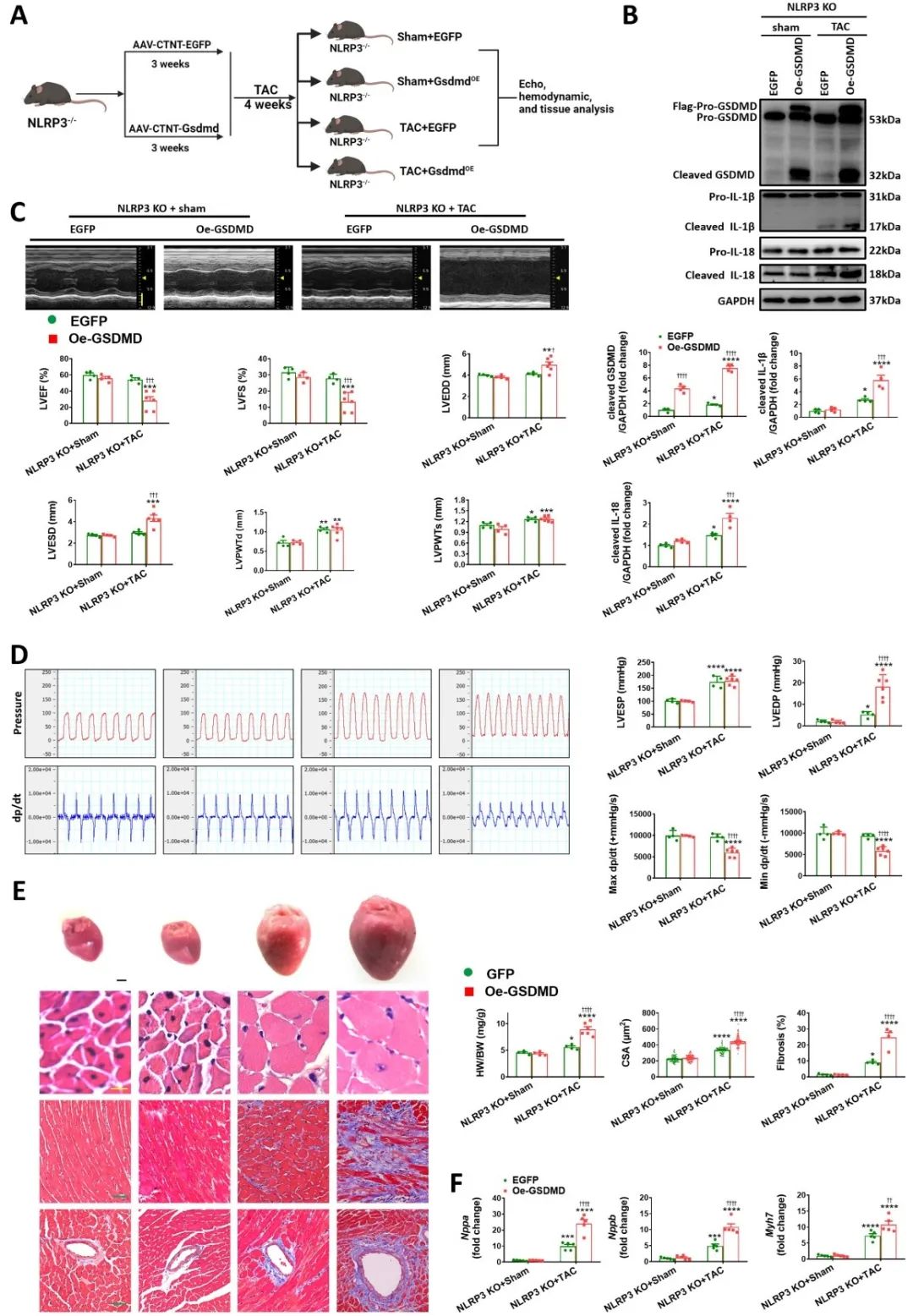
随后,对NLRP3敲除小鼠和MCC950处理的小鼠乳鼠心室肌细胞过表达Caspase-1或GSDMD,并行TAC或Ang II刺激,发现直接(GSDMD过表达)或间接(Caspase-1过表达)促进焦亡均可抵消NLRP3敲除带来的心脏保护作用。
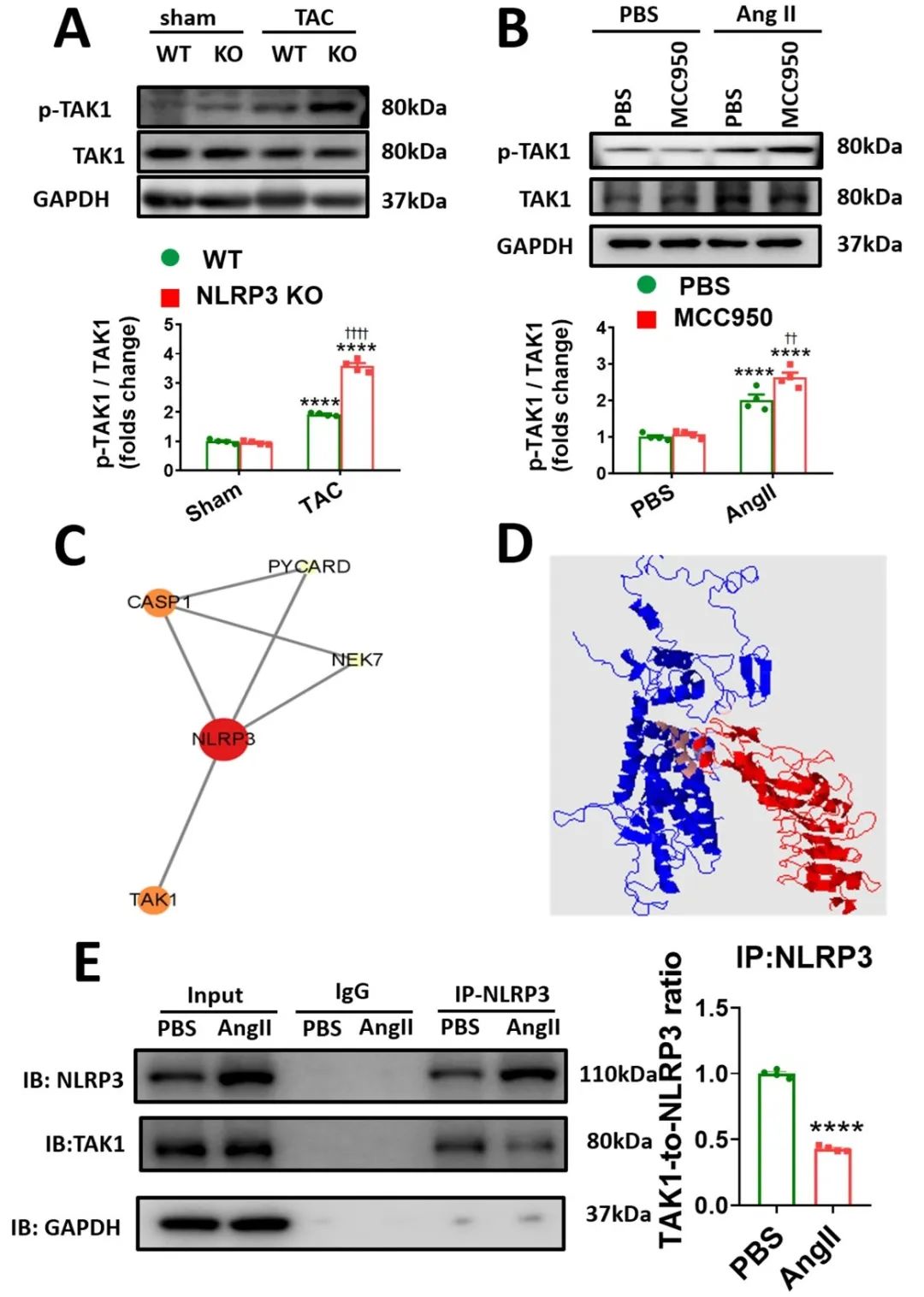
机制研究发现,压力超负荷激活TAK1,且NLRP3缺失会进一步激活TAK1。随后,通过蛋白质互作网络分析和免疫共沉淀实验确认NLRP3-TAK1存在互作。
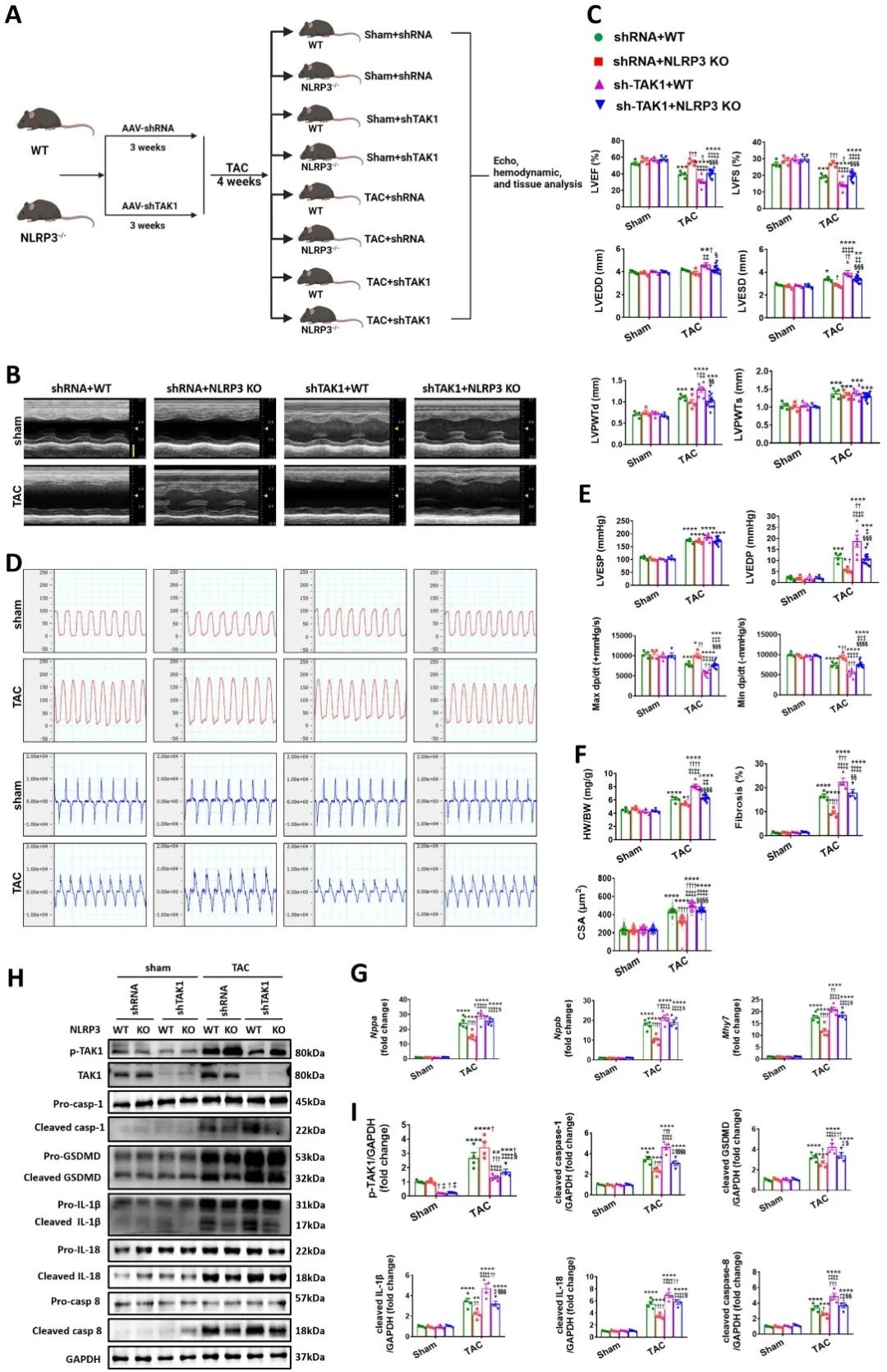
最后,在野生型和NLRP3敲除小鼠中,心脏特异性TAK1敲除加剧了心脏重构,并伴有严重的焦亡现象,进一步证明NLRP3-TAK1信号通路维持心肌细胞焦亡和肥厚稳态的重要作用。
综上,本研究较全面地探讨了NLRP3/Caspase-1/GSDMD介导的心肌焦亡事件链在压力超负荷心肌肥厚中的作用,从炎性坏死与肥大反应互作失衡的新视角提出了触发压力超负荷心脏重构的新机制,有望解决长期以来关于NLRP3和TAK1对心肌肥厚作用的争议,为心肌肥厚的精细和精准化调控提供重要参考。
复旦大学附属中山医院邹云增教授和吴剑副研究员为该文章共同通讯作者;复旦大学附属中山医院和生物医学研究院为第一署名单位。复旦大学附属中山医院李璇博士后、同济大学附属东方医院游洁芸副主任医师、贵州医科大学附属医院代方杰主治医师、复旦大学附属中山医院副研究员王时俊为共同第一作者。
复旦大学附属中山医院葛均波院士、任骏教授、孙爱军教授、杨向东教授,同济大学生命科学与技术学院/东方医院项耀祖教授、广东省呼吸疾病研究所汤海洋教授,以及加拿大皇家科学院院士、约克大学/多伦多大学Peter Backx教授对本文工作给予重要指导。该研究获得国家自然科学基金、上海市科委、上海市浦东新区卫健委等来源的基金项目资助。
原文链接:
https://www.jacc.org/doi/10.1016/j.jacbts.2023.05.008

