亨廷顿病(HD):偏爱青壮年人的“舞蹈”
时间:2023-08-25 16:06:15 热度:37.1℃ 作者:网络
亨廷顿病(HD)是一种神经退行性疾病,具有广泛的神经精神临床谱,可能涉及运动障碍(主要是舞蹈病)、痴呆和行为或精神表现的不同组合。HD是一种由位于4号染色体的亨廷顿蛋白基因(HTT)中CAG三核苷酸重复扩增引起的多聚谷氨酰胺疾病。它以常染色体显性模式遗传。CAG的正常重复范围长度在10到35之间。36到39个重复的外显率范围较低,而40个或更多重复的患者最有可能表现出这种状况。
此外,重复长度和发病年龄之间有很强的负相关。
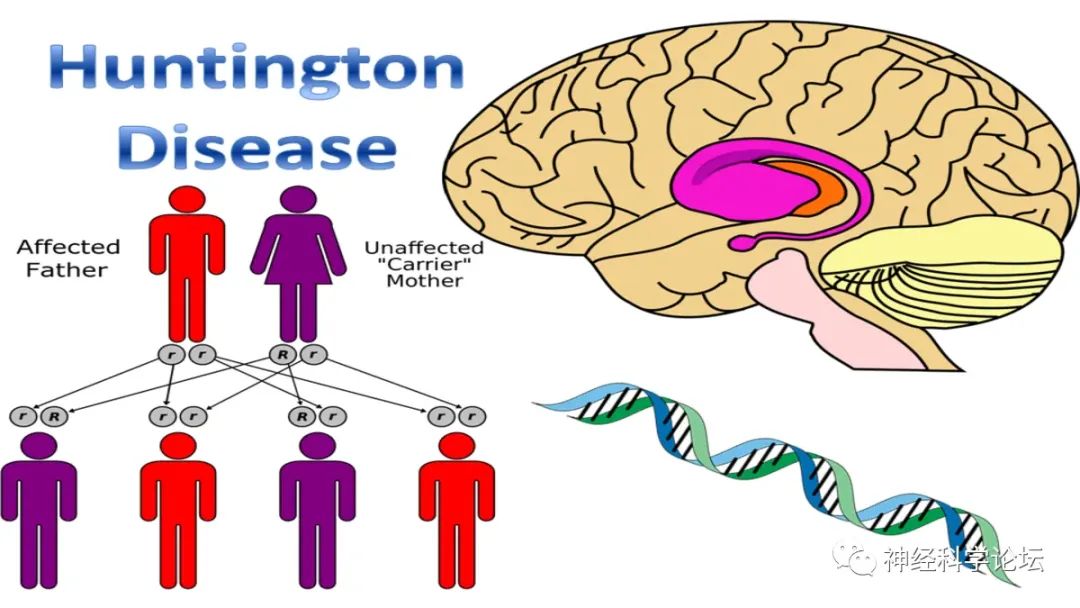
亨廷顿病(Huntington's disease,HD)是一种常染色体显性遗传性神经系统变性疾病,典型症状包括舞蹈样不自主运动、认知障碍及精神行为异常三联征。亨廷顿病是由位于4号染色体上的Huntingtin (HTT)基因1号外显子的CAG重复序列异常扩增所致。大约在1842年,Waters报道了第一个患有现在被认为是亨廷顿舞蹈症的人的病例。
然而,直到1872年,乔治·亨廷顿提出并解释了这种情况,它才被授予亨廷顿舞蹈病的称号。这是一种发生在中年的神经疾病,会在家庭中代代相传。它的特征是健忘、心理问题和无意识的合唱手势。它的名称多年来保持不变,直到20世纪90年代,亨廷顿氏症在大量非运动性主诉后被正式定义。亨廷顿氏病(HD)基因于1993年被鉴定,就在1983年第4号染色体被定位后不久。在那段时间,人们对HD和其他神经认知疾病的关注度大幅上升。然而,由于其单基因起源和完全外显率,HD是最可治愈的神经退行性疾病之一。这要归功于在过去十年中开发了新的治疗策略,其可以特异性靶向亨廷顿蛋白基因并抑制破坏性突变亨廷顿蛋白的合成。
亨廷顿病在各种族人群均有报道,以高加索人种多见,患病率为(10.6~13.7 ) /10万;亚洲地区罕见,日本、中国台湾和香港地区患病率约为(0.1~0.7 ) / 10万,中国大陆目前缺乏流行病学数据。欧洲人后裔的发病率是东亚人的10-100倍。根据1985年至2010年发表的研究,HD的总发病率估计为每100,000人年0.38人,全球总患病率为每100,000人年2.71人。
最近的研究强调,在一些地区,流行率有所上升。尽管对不同人群中的差异和增加率有合理的解释,如不同HTT基因单倍型的识别、医疗保健的可及性、对疾病相关污名的不同态度、迁移和HD聚集区的识别,但具体的决定因素仍有待阐明。
由突变的亨廷顿蛋白基因引起的功能障碍和神经元死亡涉及许多途径。基因座1突变亨廷顿蛋白(mHTT)片段的直接影响,mHTT产生异常聚集物的趋势及其对细胞稳态、神经元转运、转录、翻译、线粒体功能和突触功能的影响。纹状体中多刺神经元特别容易受到这种副作用的影响。纹状体病理学分两个阶段发展。第一阶段的特征是间接途径的中棘神经元(MSNs)的缺失,这导致运动过度表型,第二阶段的特征是直接途径的中棘神经元的缺失,这导致运动不足特征。未知因素可能导致了MSNs选择性易感性的间接途径;然而,D2多巴胺受体可能发挥作用,因为它们与亨廷顿舞蹈症的发病机制有关,并间接表达,但不直接表达MSNs。其他推测还包括脑源性神经营养成分的损耗、来自皮质-纹状体评估的谷氨酸兴奋毒性以及重复相关非ATG翻译蛋白的有害后果。

HD平均发病年龄在35-45岁之间。这种情况通常持续16-18年。随着病情的发展,对日常工作的依赖性增加,最终导致死亡。亨廷顿病的初始运动指征可能是肌张力障碍。抽搐是另一种不受欢迎的运动,尽管它们相对不常见。小脑主诉可能表现不稳定,与近视和近视很相似。在许多情况下,运动被比作“醉酒”或经历小脑共济失调。区分舞蹈症和共济失调步态是非常具有挑战性的。不论发病年龄,HD都是一种慢性、缓慢进行性疾病。临床发病后的平均存活时间为10~20年,部分患者可存活30~40年。
HD主要表现为不自主的舞蹈样动作和严重的精神障碍。随着病情的发展,患者会丧失说话、思考甚至吞咽的能力。伴随着病情逐渐连续进展,HD可分为几个概括预期临床病程的阶段。
-
早期:患者通常功能正常且可以独立完成日常生活的大多活动,往往可以生活自理、可以工作和驾驶,但是存在HD症状,如轻微的不自主运动、轻微的不协调、难以完成复杂的智力任务,以及一定程度的易激惹、脱抑制或抑郁。患者可能有缓慢的眼球自主运动,尤其是较年轻的患者。
-
中期:患者开始无法独立工作、驾驶或管理自己事务[61]。大多数情况下,患者进食、穿衣和个人护理需要一些协助。认知减退加重,解决问题的能力下降。患者通常有舞蹈病症状加重和自主运动困难。行走和平衡受损可能导致跌倒。其他问题可能包括吞咽障碍和体重减轻。
-
晚期:认知和运动功能缓慢但持续不断的恶化可导致严重并发症和早期死亡。HD晚期,患者无法完成日常生活的一切活动,需要24小时监督和护理。自主运动控制的丧失加重;余下的少数自主运动往往是大幅度投掷运动,可能引起跌倒损伤或肢体创伤。大多数患者卧床不起。由于吞咽困难,患者可能需要放置喂食管,以维持足够的营养。HD晚期患者通常不能言语,但可能保留一定理解力。
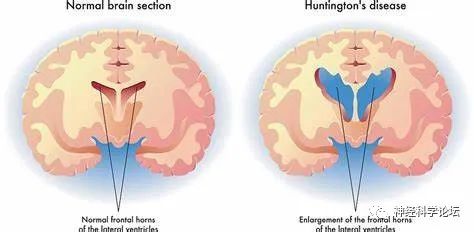
虽然HD患者都需要24小时照护,但晚期HD患者的疾病表现可有显著差异。例如,一些患者有明显、严重的行为障碍,一些有严重的舞蹈病和步态障碍但没有明显的行为障碍,而另一些意识不到自己的症状。
HD临床是通过评估家族史、个人史、神经和精神检查以及基因检测做出的临床诊断。目前的分类方案将HD细分为症状前、前驱和显性HD。症状前HD包括CAG扩张但目前没有HD相关体征或症状的个体。前驱HD包括CAG扩张的个体,这些个体在检查中具有非特异性或可能的运动异常以及细微但清晰的认知变化。显性HD包括CAG扩展的个体,其运动异常的置信度大于90%,加上轻微或主要的神经认知变化,或运动异常的概率大于99%,但认知不变。未进行HD基因检测但临床上怀疑有HD诊断的个体将被归类如下:有HD风险,但不明显;临床前驱HD;和临床表现为HD。
根据统一的亨廷顿氏病评定量表、总运动评分和诊断置信度评分,运动损伤的开始是确诊亨廷顿氏病所必需的,因为这需要证明家族史或甚至是阳性基因测试。4分表示运动性发作或“明显的”亨廷顿舞蹈症。这个范围从零到四。然而,这种疾病的先兆期(可在明显疾病发作前11-16年发现)是指轻度的运动、智力和心理缺陷。
HD是第一个被描述的三核苷酸疾病,也是第一个在症状出现前可能被诊断的常染色体显性疾病。自1983年和基因定位以来,对该疾病的了解显著增加,这对于通过发现新的分子靶点来改善患者的生活质量和改善治疗策略是必要的。在过去的20年里,人们对疾病的认识和对病人的治疗都有了很大的提高。人们常常忽略在出现症状之前在危险和发展阶段度过的许多年,也称为先兆期,因为典型的病程甚至超过17年。无论是患者还是家庭都会在他们的余生中受到HD的影响。
在20世纪90年代中期基因检测出现之前,流行病学研究主要依赖于运动临床谱以及类似表现的阳性家族史。这种方法可能由于包括了高清拷贝而高估了实际比率。另一方面,由于缺乏明确的传播模式,估计占病例7.1%的新基因突变可能被忽略。HTT基因的鉴定、定位和随后的克隆促进了分子诊断,允许预测性检测,并可能增加非典型表现形式的检出率。
此外,风险人群和预测性测试计算的采用作为临床实践和研究目的识别病例的措施出现。然而,分子检测最初的可用性是有限的,即使在卫生保健系统健全的管辖区也是如此,流行病学研究的诊断纳入标准在很大程度上仅基于临床依据。自从之前发表的关于全球HD患病率和发病率的系统综述和荟萃分析以来,文献中出现了许多新的流行病学报告(特别是自2016年以来),扩大了对确诊病例的确认性基因检测的利用。
当基因被发现时,如何治疗这种可怕疾病的坚实理由首次出现,提供了新的研究途径和建模可能性。已经有几种对症疗法可供选择。然而,仍然需要改进的修饰药物。HD通过各种过程,包括蛋白沉积、转录和细胞功能的损伤,以及突变蛋白的直接毒性,突变的亨廷顿蛋白在细胞水平上引发神经元功能障碍和损失。随着疾病的恶化,大脑与纹状体最初的宏观变化一起受到影响。由于目前很少有药物可以改变疾病的进程,姑息疗法和症状控制是治疗的基础。通过家族的早期检测和阻断其病程来识别可能有助于预防HD表现的有效生物标志物也是至关重要的。
专注于理解导致HTT基因突变的潜在分子机制的研究非常有希望,旨在找到治疗HD的方法。然而,目前对HD的治疗仍然有限。所应用的治疗集中在症状的治疗上,因为防止疾病发作和减缓疾病发展的神经保护疗法还不可用。近年来,随着疾病的发展,对细胞病理学和大脑总体结构变化的研究取得了巨大进展。越来越多的证据支持炎症在神经退行性疾病中起着关键作用,并刺激了免疫治疗策略在调节神经炎症性疾病中的应用。一些潜在免疫调节药物的临床前和临床试验已在HD中进行,如米诺环素和大麻素类药物、拉喹莫德、TNF-α抑制剂、抗SEMA4D单克隆抗体、神经节苷脂等。HD进展期可持续10年或更长时间,取决于治疗/护理水平。不动的并发症(例如,吸入性肺炎和其他感染)导致患者在发病后10-40年死亡。
参考文献
Medina A, et al. Prevalence and Incidence of Huntington's Disease: An Updated Systematic Review and Meta-Analysis. Mov Disord. 2022 Dec;37(12):2327-2335. doi: 10.1002/mds.29228.
Andhale R, et al. Huntington's Disease: A Clinical Review. Cureus. 2022 Aug 27;14(8):e28484. doi: 10.7759/cureus.28484.
Jia Q, et al. Neuroinflammation in Huntington's disease: From animal models to clinical therapeutics. Front Immunol. 2022 Dec 22;13:1088124. doi: 10.3389/fimmu.2022.1088124.
Palaiogeorgou AM, et al. Recent approaches on Huntington's disease (Review). Biomed Rep. 2022 Nov 21;18(1):5. doi: 10.3892/br.2022.1587.
Dai Y, et al. A comprehensive perspective of Huntington's disease and mitochondrial dysfunction. Mitochondrion. 2023 May;70:8-19. doi: 10.1016/j.mito.2023.03.001.

