中华医学会第一届全国罕见病学术年会(CSRD 2023):法布雷病诊断和治疗进展
时间:2023-09-24 11:13:40 热度:37.1℃ 作者:网络
2023年9月22-23日,由中华医学会、中华医学会罕见病分会主办的中华医学会第一届全国罕见病学术年会(CSRD 2023)于北京国际饭店隆重召开。本次大会邀请国内外罕见病领域知名专家进行专题报告、指南解读、病例讨论、大会发言及论文交流等内容。
本次大会,来自四川大学华西医院的陈玉成教授为我们分享《法布雷病诊断和治疗进展》议题。梅斯医学整理重点分享给各位。

法布雷病是一种罕见的X连锁遗传溶酶体贮积症
法布雷病是一种罕见的X连锁遗传溶酶体贮积症,正常人群中预估患病率为1/100000。国外报道,新生儿中法布雷病发病率约为1/1250~1/8882。

法布雷病临床表现多样,可累及多种器官、系统
法布雷病临床表现多样,常为神经、肾脏、心脏、皮肤、胃肠道、眼等多脏器受累。患者多在青少年时期出现症状,并随病程进展而逐渐加重。其中,肾脏、心脏、脑是后期主要受累脏器。
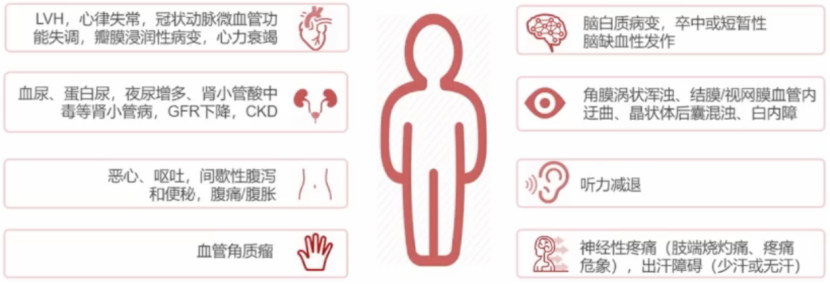
法布雷病按临床表现分为经典型和迟发型
国外文献报道迟发型发病率比经典型高出10倍,据上海交通大学医学院附属瑞金医院统计资料显示,国内目前诊断的患者中66.1%男性患者为经典型,75%女性患者为迟发型法布雷病的临床分型。

法布雷病是一种进展性疾病,最终会导致多器官功能衰竭,甚至过早死亡
男性患者寿命预期减少约15~20年,女性患者寿命预期减少约6~10年。

为了规范法布雷病的诊疗,近年来一系列指南共识应运而生

法布雷病目前诊断方法
由于法布雷病缺乏特异性症状,因此,法布雷病的诊断需结合临床表现、酶活性、基因检测、生物标志物等多项指标。
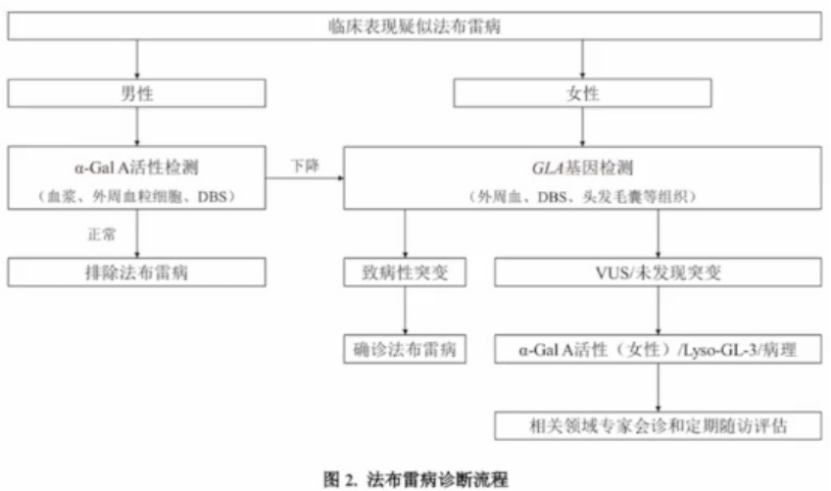
a-GalA活性检测
用于男性患者的诊断。
样本来源:血浆、外周血粒细胞、干血纸片(DBS)
诊断要点:
- 男性:严重下降或缺失,提示患有法布雷病
- 女性:大多数女性患者在正常范围,因此需要基因检测来明确诊断

基因检测
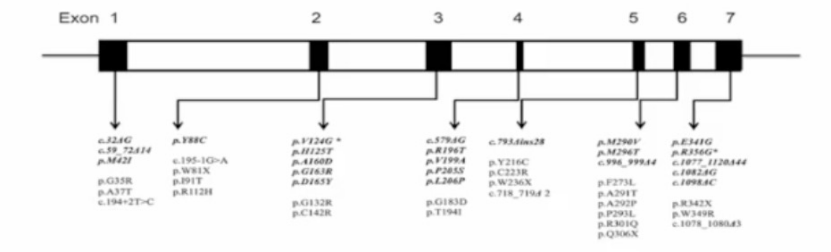
用于明确诊断,确定基因突变类型,判断临床表型和家系筛查。
在73例中国法布雷病患者中发现47种突变,包括23种新突变。
47种突变包括:
- 错义突变 (66%)
- 无义突变(8.5%)
- 剪接突变(4.3%)
- 移码突变(21.2%)
生物标志物检测
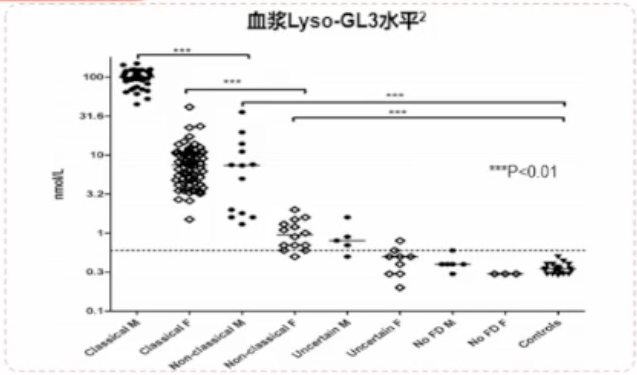
血浆Lyso-GL3检测用于:
- 辅助诊断
- 区分经典型和迟发型
- 对VUS且α-Gal A活性正常(主要女性患者)或降低的男性/女性患者
- 人群筛查
样本来源:血浆、外周血粒细胞、干血纸片(DBS)
诊断要点:
- 男性:可监测疾病严重度和进展
- 女性:假阳性率偏高,诊断值的参考范围尚待更多循证医学证据支持
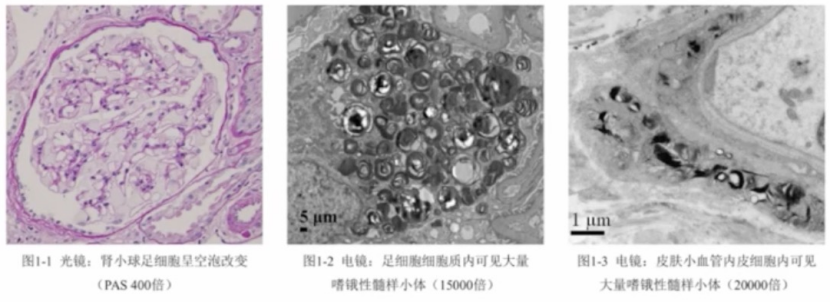
病理活检
用于辅助诊断。
标本来源:肾脏、心脏、皮肤或神经组织
诊断要点:
光镜下:相应组织细胞呈空泡改变
电镜下:相应的组织细胞胞质内充满嗜饿性“髓样小体”
加拿大指南推荐的法布雷病诊断标准
经诊断评分为≥3分的患者诊断为法布雷病。

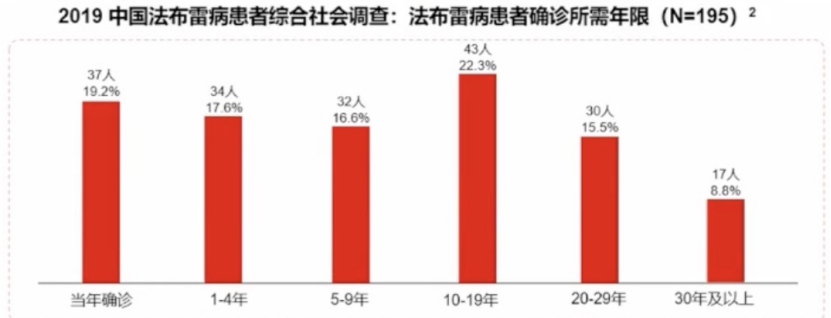
目前我国法布雷病误诊率高,平均确诊延迟约14年以上
法布雷病患者常存在诊断延迟,主要原因为疾病较为罕见(临床医师对其缺乏认知)以及临床症状的多样性和非特异性。
2019年的中国法布雷病调查显示,法布雷病患者误诊率达69.7%,患者人均花费14年以上才能得到确诊。
如何提高法布雷病的诊断率?——特定人群筛查
家系筛查、高危人群筛查和新生儿筛查,可极大的提高法布雷病患者的阳性诊断。
1.家系筛查:对发现新的法布雷病患者有重要意义,帮助患者及早开始治疗。
2.高危人群筛查:可提高法布雷病的诊断率,及时采取有效治疗措施避免严重并发症发生。
- 肾脏疾病:CKD、透析
- 心脏疾病:LVH、HCM
- 神经系统疾病:缺血性脑卒中
3.新生儿筛查:在临床症状和体征出现之前及早发现疾病,及早开始治疗,避免严重并发症或死亡发生。
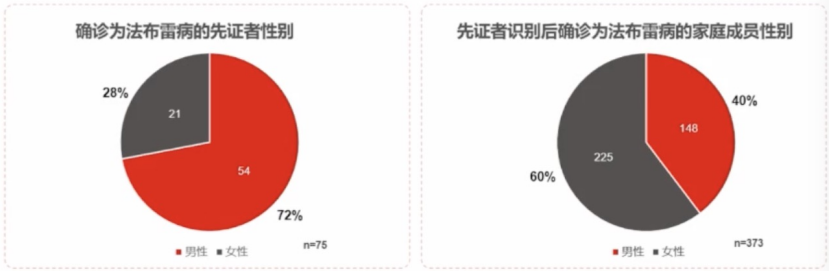
家系筛查:对已确诊法布雷病患者进行家系筛查
在75例先证者家系筛查中共确诊法布雷病373例,分析发现,平均每个先证者有5个家庭成员被诊断为法布雷病。
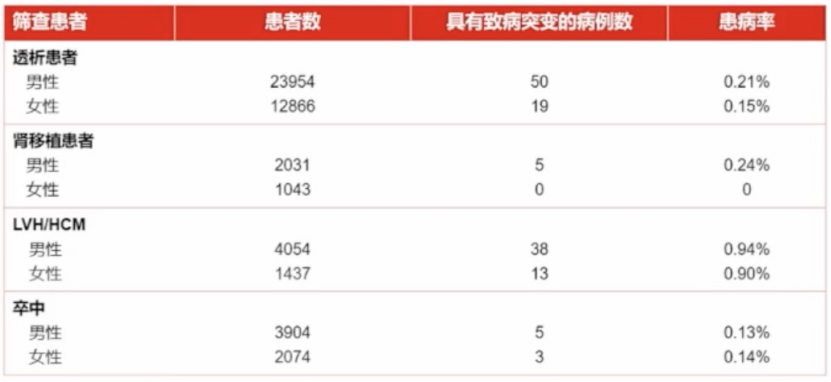
高危人群筛查:全球筛查数据
该研究分析了1995-2017年期间法布雷病在析、肾移植、心脏和卒中患者中的筛查报告数据通过对报告GLA突变的研究重新分析致病突变以描述法布雷病的患病率。共纳入63项研究。
高危人群筛查:中国筛查数据
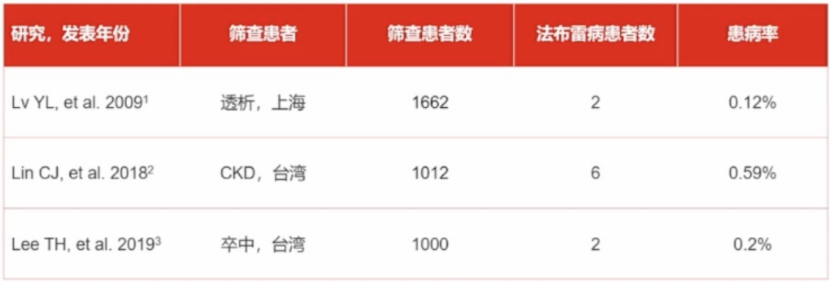
华西医院左心室肥厚表型患者筛查
基于前瞻性HCM临床磁共振队列进行Fabry筛查,连续性纳入华西医院602例HCM患者,筛查发现Fabry患者11例,检出率1.8%。
法布雷病的治疗方法
法布雷病的治疗目标:延缓疾病进展,改善生活质量,降低相关并发症的发病率,延长患者生存期。指南提倡对法布雷病进行个体化管理,疾病特异性治疗和对症治疗相结合,多学科团队共同管理。

酶替代疗法(ERT)为法布雷病治疗的基石
ERT作为外源性补充基因重组的α-Gal A,替代患者体内缺失的α-GalA,从而促进GL-3的分解减少GL-3和Lyso-GL-3在器官组织的贮积。

目前国内获批的两种ERT药物

各国指南对于ERT起始治疗指征的推荐——儿童患者

各国指南对于ERT起始治疗指征的推荐——成人患者

对症治疗
对症治疗主要是针对各脏器受累情况,给予相应的对症治疗。

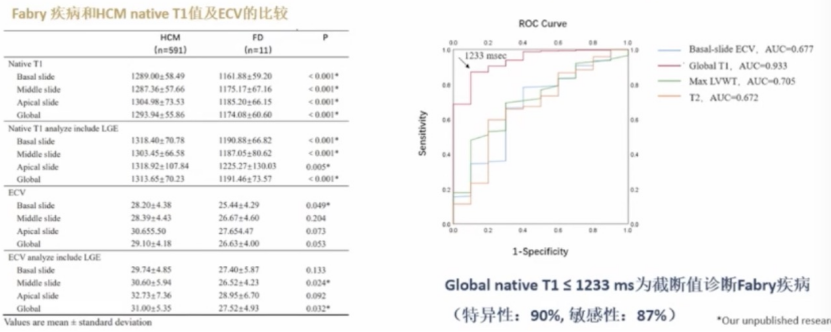
分子伴侣疗法可矫正缺陷酶,恢复部分酶活性

分子伴侣疗法可显著减少法布雷病患者肾脏足细胞GL-3贮积
目前,临床使用较多的是米加司他,已在加拿大、欧洲等国家上市,暂未在中国上市,仅适用于部分错义突变的法布雷病患者。米加司他分子伴侣疗法是一种口服治疗法布雷病的方法,可通过促进突变的α-Gal A蛋白正确折叠,提高其稳定性从而增加酶活性。
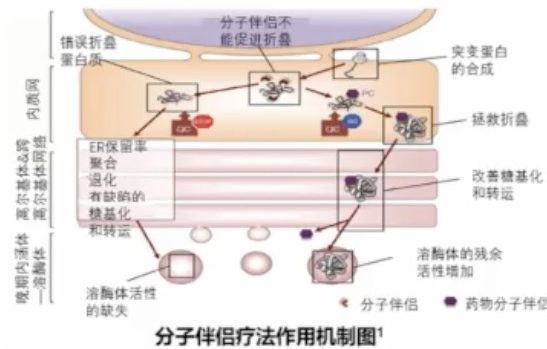
一项随机、双盲、开放标签的试验指出:米加司他治疗法布雷病6个月后肾脏足细胞GL-3贮积显著下降,肾脏血管内GL-3水平较安慰剂组显著下降,胃肠道症状有所改善。
SRT抑制底物积累

SRT是一种能够透过血脑屏障的口服葡糖神经酷胺合成酶(GCS)抑制剂,可经口服特异性治疗法布雷病,限制代谢产物GL-3的形成,暂未在中国上市。

SRT可显著降低法布雷病患者血浆GL-3及Lyso-GL-3水平
一项国际多中心、开放标签、单臂研究(2a期临床试验)显示:接受Venglustat治疗26周后,血浆GL-3、Lyso-GL-3水平较基线分别降低47.1%(P<0.001)、30.8%(P=0.0036);延长治疗至156周,两者较基线分别进一步降低77.5%、52.5%。
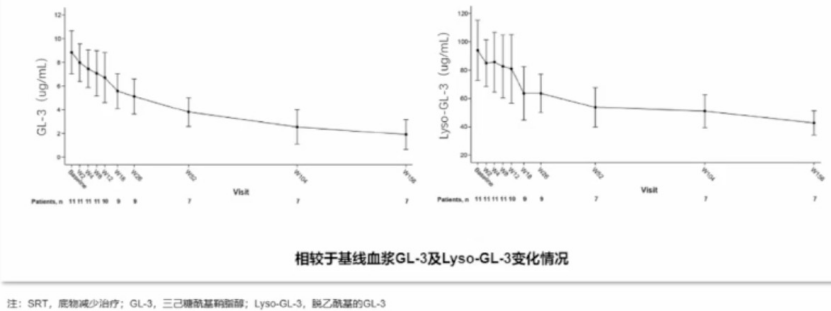
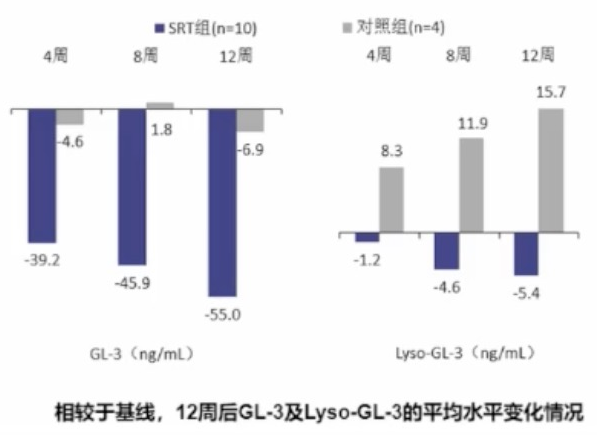
一项单中心、开放标签、随机研究显示:接受Lucerastat治疗12周后,GL-3及Lyso-GL-3水平较基线分别下降-55.0ng/mL和-5.4ng/mL(P均<0.0001)。
基因治疗可降低法布雷病患者血浆和尿液的GL-3及Lyso-GL-3水平
基因治疗技术尚未获批用于法布雷病,但目前已开展用多种载体(逆转录病毒、慢病毒、腺病毒、腺相关病毒和非病毒载体)进行体内、体外基因治疗的临床研究,暂未在中国上市。
主要通过载体介导将正常GLA基因转移至法布雷病患者体内,使患者机体产生具有正常活性的α-Gal A。
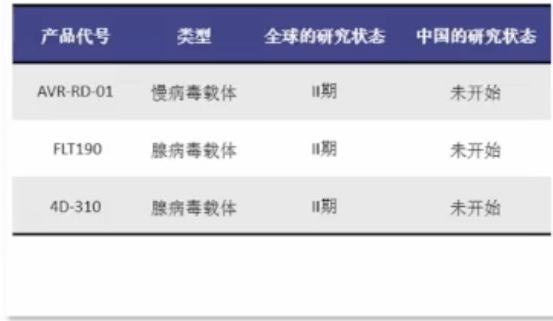
一项多中心、单臂试验显示:5例经典型男性法布雷病患者接受基因治疗1周后酶活性接近正常水平,血浆和尿液中GL-3及Lyso-GL-3水平降低。

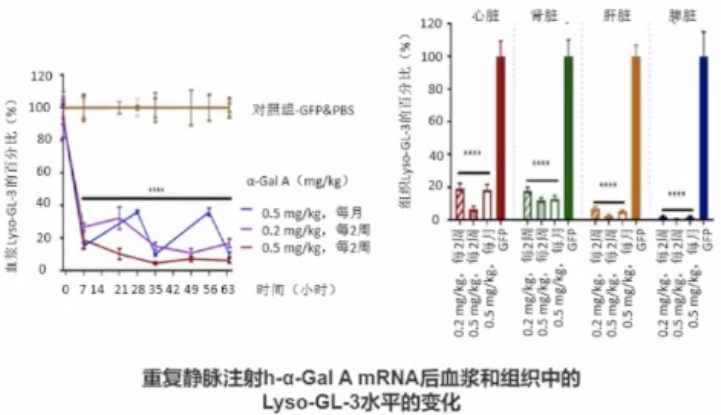
mRNA治疗可减少法布雷病小鼠Lyso-GL-3和GL-3的底物贮积
小鼠和非人类灵长类动物实验显示,mRNA疗法被证明可以降低血浆、肾脏和心脏中GL-3和Lyso-GL-3水平。目前该治疗方法在人类中应用还需进一步研究,暂未在中国上市。
mRNA治疗不存在插入突变的风险,但效果短暂,需重复给药,该疗法通过脂质纳米材料包裹α-Gal A mRNA进入主要产生α-Gal A的肝细胞,在肝细胞中产生酶并分泌到循环中。

动物研究显示在血浆、心脏、肾脏、肝脏和脾脏中观察到Lyso-GL-3水平减少。在接受不同剂量h-α-Gal-A mRNA的动物组织中,发现Lyso-GL-3水平均降低80%以上。
总结
法布雷病是一种罕见的X连锁遗传性疾病,可累及多种器官、系统,最终导致心脏/肾脏等重要器官疾病和早亡。法布雷病的诊断需结合临床表现、酶活性、基因检测、生物标志物等多项指标,确诊需依靠酶学检查和基因检测。法布雷病在肾脏疾病(慢性肾脏病、透析)、心脏疾病(左心室肥厚、肥厚型心肌病)、神经系统疾病(缺血性脑卒中)人群中患病率较高,对这类高危人群进行筛查,可提高法布雷病患者的诊断率。ERT为法布雷病患者的治疗基石,对法布雷病患者治疗效果显著,患者及早启动治疗获益更大。法布雷病新的治疗正在开发中,将可能进一步减少重要脏器损害。

