JMC:胰高血糖素样肽-2受体强效和选择性D构型多肽激动剂的设计
时间:2023-09-29 11:23:11 热度:37.1℃ 作者:网络
与小分子药物相比,多肽与靶点的亲合力强、特异性高、副作用小,但多肽类药物的物理化学性质不稳定、容易被蛋白酶水解、半衰期较短、清除速率快、不容易透过细胞膜,并且大多数药物不能口服。由于体内蛋白酶所能识别的氨基酸为天然的L-氨基酸,因此用D-氨基酸代替L-氨基酸可有效防止蛋白酶对多肽的降解。多伦多大学Philip M. K.小组针对胰高血糖素样肽-2受体 (GLP-2R) 设计了三种D-GLP-2激动剂,它们能激活胰高血糖素样肽-2 受体 (GLP-2R), 进而增加环磷酸腺苷 (cAMP) 的表达,而不激活胰高血糖素样肽-1 受体 (GLP-1R)。所有D-GLP-2激动剂均能以时间和浓度依赖性方式增加蛋白激酶B磷酸化 (p-AKT) 的表达水平,同时D-GLP-2激动剂对蛋白酶降解具有较强的稳定性,不仅可以降低治疗所需的剂量,而且还可以增强其在肠道的吸收以及在治疗炎症性肠病方面的潜在应用。相关工作以“Computational Design of Potent and Selective d-Peptide Agonists of the Glucagon-like Peptide-2 Receptor”为题发表在美国化学会出版的药物化学核心期刊Journal of Medicinal Chemistry上 (J. Med. Chem, 2023, 66 (15): 10342-10353)【1】。
胰高血糖素样肽2 (glucagon-like peptide-2, GLP-2) 是一种肠道多肽类激素,具有多种肠道效应,包括刺激肠黏膜生长、促进营养物的消化和吸收、提高肠屏障功能、抑制胃能动性和胃酸分泌等。GLP-2通过作用于GLP-2受体 (GLP-2 receptor,GLP-2R) 来发挥生物学作用。GLP-2R是G蛋白偶联受体超家族成员之一,具有7个跨膜结构域,与胰高血糖素、GLP-1 和葡萄糖依赖的促胰岛素多肽的受体高度同源。近期,报道的GLP-2R与GLP-2和Gs异源三聚体配合物的电镜结构显示GLP-2R与GLP-2具有独特的肽识别机制,为开发选择性GLP-2R激动剂提供了机会。
然而,二肽基肽酶4 (DPP-IV) 可迅速降解GLP-1和GLP-2,限制了GLP-2肽类药物的治疗应用。由于天然蛋白酶只能识别和切割由L-氨基酸组成的蛋白质,因此,由D-氨基酸组成的肽具有高度的抗蛋白酶水解能力。由于D-氨基酸侧链取向的完全改变,简单地用D-氨基酸替换L-氨基酸通常是无效地。为解决这一问题,作者将PDB文件中的所有非蛋白分子去除后,将整个文件沿X轴翻转得到镜像版本,并命名为D-PDB,然后去除蛋白质的非螺旋部分,并拆分蛋白结构得到若干个螺旋结构,并以此建库 (图1)。随后,作者将GLP-2螺旋结构分为三个重叠的片段,并在每个片段中挑选三个对结合有显著贡献的热点残基。然后,根据热点残基的构象扫描D-PDB数据库来获得包含D-氨基酸短肽片段。最后,将最佳匹配的肽段组合成D-GLP-2肽类似物。值得注意的是,组装完成后,GLP-2和D-GLP-2的热点残基之间保持了高度配对。
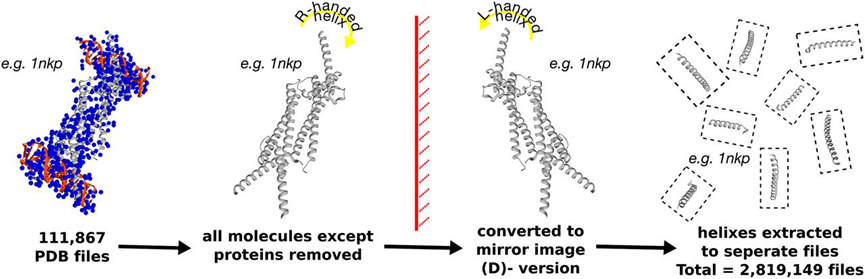
图1: D-PDB数据库构建流程图
作者通过建模模拟GLP-2R受体的结构,并通过300 ns MD模拟评估D-GLP-2的结合稳定性。结果显示D-GLP-2和L-GLP-2占据着相同的结合位点。随后,作者使用MM/GBSA进行丙氨酸扫描,以研究D-GLP-2中不同残基对GLP-2R结合亲合力的贡献。正如预期的那样,D-肽类似物中的大多数残基与GLP-2的热点残基一样对于结合具有显著的贡献,但E33对结合展现出负影响 (图2A)。作者猜测是因为E33位于GLP-2R TM结构域的E398残基附近导致的负贡献。随后,作者使用Crooks Gaussian interp (CGI) 方法来评估E33A、E33F和E33Y点突变对D-GLP-2与GLP-2R亲合力的影响。自由能计算表明,所有的突变都会增加结合亲合力。考虑到D肽与L-GLP-2具有相反的取向,作者假设在C端残基上引入一个肼基 (NH-NH2)可以提高活性得到D-GLP-2 E33A Hydrazide (图2B)。
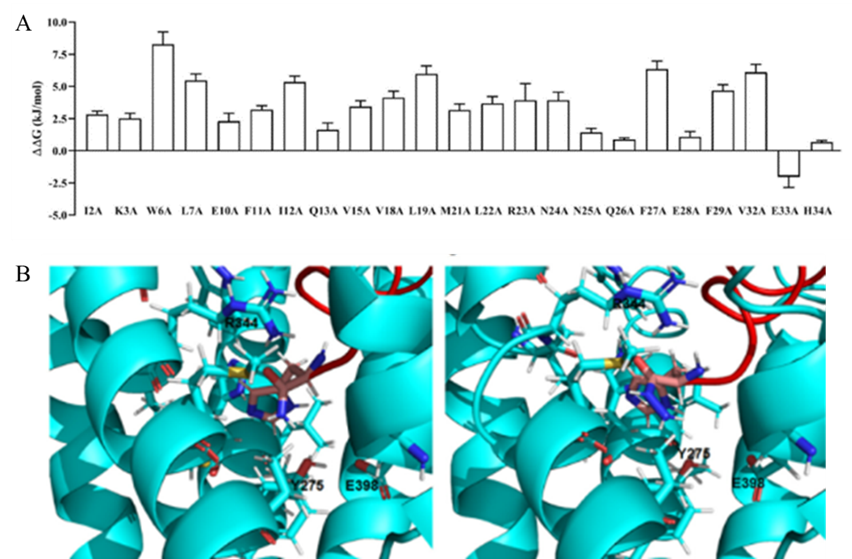
图2: 基于计算的D-GLP-2的重新设计。(A) 使用MM / GBSA方法对GLP-2R + D-GLP-2复合物的MD模拟进行计算丙氨酸扫描;(B) D-GLP-2类似物与GLP-2R的对接图
在完成D-GLP-2肽设计后,作者首先通过圆二色谱(CD) 鉴定肽的二级结构,结果显示所有D-肽在溶液中均能保持螺旋状。接下来,作者创建稳定表达GLP-2R的HEK293细胞系,并通过TR-FRET以确定L-GLP-2和D-GLP-2与GLP-2R的结合能力。结果显示:L-GLP-2的EC50 = 40.4 nM,而D-GLP-2、E33A、E33A hydrazide的EC50分别为1417、414和226 nM (图3A)。同样,作者也对GLP-1R进行相应的测试,发现无论是D-GLP-2还是L-GLP-2均不能激活GLP-1R (图3B),表明GLP-2R具有独特的配别识别功能。随后作者通过β-半乳糖苷酶互补法测量了D-GLP-2类似物和L-GLP募集β-arrestin 2的能力,发现D-GLP-2类似物招募β-arrestin 2的能力明显低于L-GLP-2。最后,作者研究了D-GLP-2类似物激活GLP-2R导致的下游信号通路的影响,发现所有肽诱导相似的GFP的表达,而D-GLP-2 E33A hydrazide诱导p-AKT表达的能力是L-GLP-2的2.28倍 (图3C, D)。酶抗性是衡量肽类药物稳定性的重要指标之一,作者最后通过DPP-IV和蛋白酶K两种蛋白酶评估了D-GLP-2类似物对水解酶的抵抗力。结果显示所有D-GLP-2类似物在DDP-IV中孵育2.5h后仍能保持完整,且在水解能力更强的蛋白酶K中表现出比L-GLP-2-2G更强的酶抗性 (图3E)。
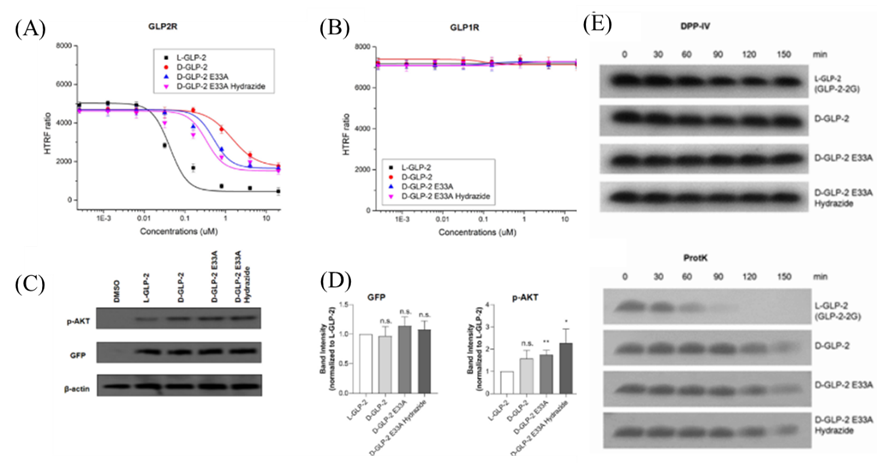
图3: D-GLP-2类似物的实验验证。(A, B) TR-FRET实验测试结果;(C, D) D-GLP-2 类似物和天然 L-GLP-2 在 10 μM 处诱导的 GFP 和蛋白激酶 B 磷酸化 (p-AKT)表达水平的WB结果图;(E) 蛋白酶处理D-GLP-2 类似物不同时间的WB结果图。
总结
该论文作者通过先前构建的D-螺旋肽数据库,筛选与L-GLP-2热点残基具有相同构象的D-多肽,得到了三种D-GLP-2激动剂,它们均能激活 GLP-2R, 进而增加cAMP的表达,而不激活GLP-1R。并且所有D-GLP-2激动剂均具有较强的蛋白酶抵抗性,降低了当前治疗所需的剂量,在增强肠道吸收和治疗炎症性肠病方面具有潜在的应用价值。同时,D-PDB匹配的方法为药物早期发现寻找稳定的先导肽类分子提供了新的方向。
参考文献
【1】 Valiente P A, Nim S, Kim J, Kim P M.Computational Design of Potent and Selective d-Peptide Agonists of the Glucagon-like Peptide-2 Receptor. J., Med., Chem., 2023, 66 (15): 10342-10353. .

