Cell:一种基于无参考文献的统计基因组算法SPLASH
时间:2023-12-15 10:46:06 热度:37.1℃ 作者:网络
基因组学现在是生物学、生态学和医学的基础,随着测序数据库的发展,利用它们进行发现的机会也在增长。如何最好地分析这些数据来揭示监管和功能?传统上,生物信息学管道从通过与参考基因组对齐来分配基因组位置开始,这种方法有许多局限性。对于研究较少的生物体,参考资料可能部分组装错误、不完整或不存在。即使在深入研究的人类基因组中,也发现研究不足的人群在当前参考文献中也缺少大量序列;这种盲点可能会加剧健康差距。
基于参考的方法不适合处理并行逻辑和重复元素(占人类基因组的约54%2),因此许多分析只是忽略了它们。它们也不适合癌症等几乎由偏离参考定义的疾病,即使在单个肿瘤内也是如此。此外,病毒和微生物基因组的巨大多样性及其不断适应使得定义一整套参考资料变得不可行。实际上,对参考的对齐是计算密集型的,限制了基因组推理的规模。
在处理基因组数据时,精确的统计分析至关重要。然而,基于对齐的方法很复杂,很难进行统计建模;即使是看似简单的任务,如调用等位基因特定表达式,也可能充满对齐期间引入的统计不准确性。基于排列的方法不是灵丹妙药;除了缓慢外,它们还可以低估虚假发现率的10倍。
解决这些问题使我们形成了一个简单、统一的范式,直接从原始测序数据中统计检测生物感兴趣的信号,而无需使用参考基因组,称之为SPLASH(统计初级对齐不可知序列寻址)。它依赖于序列变化的简单形式化(不同序列的短段,“目标”,毗邻短段恒定序列,“锚”)。SPLASH适用于无数的生物问题,这些问题可以被描述为询问一组样本中的序列分布如何变化。

2023年12月7日发表在Cell的文章,提供了SPLASH发现广泛可能性的快照,包括病毒株变异、单细胞级替代异构体和人类样本中的抗原受体多样性。本文还表明,SPLASH很容易应用于研究较少的生物:狐猴、章鱼和鳗草。这表明SPLASH有可能在许多生物问题和生物体中发现有意义的序列变异,而无需参考基因组的帮助。
研究人员引入了一个统一的范式,SPLASH(统计初级对齐不可知序列归因),它直接分析原始测序数据,使用统计测试来检测调节的特征:样本特定的序列变异。SPLASH检测许多类型的变异,并且可以有效地大规模运行。
研究结果显示,SPLASH识别了SARS-CoV-2中的复杂突变模式,在单细胞水平上发现了受调控RNA异构体,检测了适应性免疫受体的巨大序列多样性,并发现了其参考基因组中未记录的非模型生物的生物学:鳗草(一种受气候变化影响的海洋植物)中的地理和季节性变异以及硅藻关联,以及章鱼的组织特异性转录物。
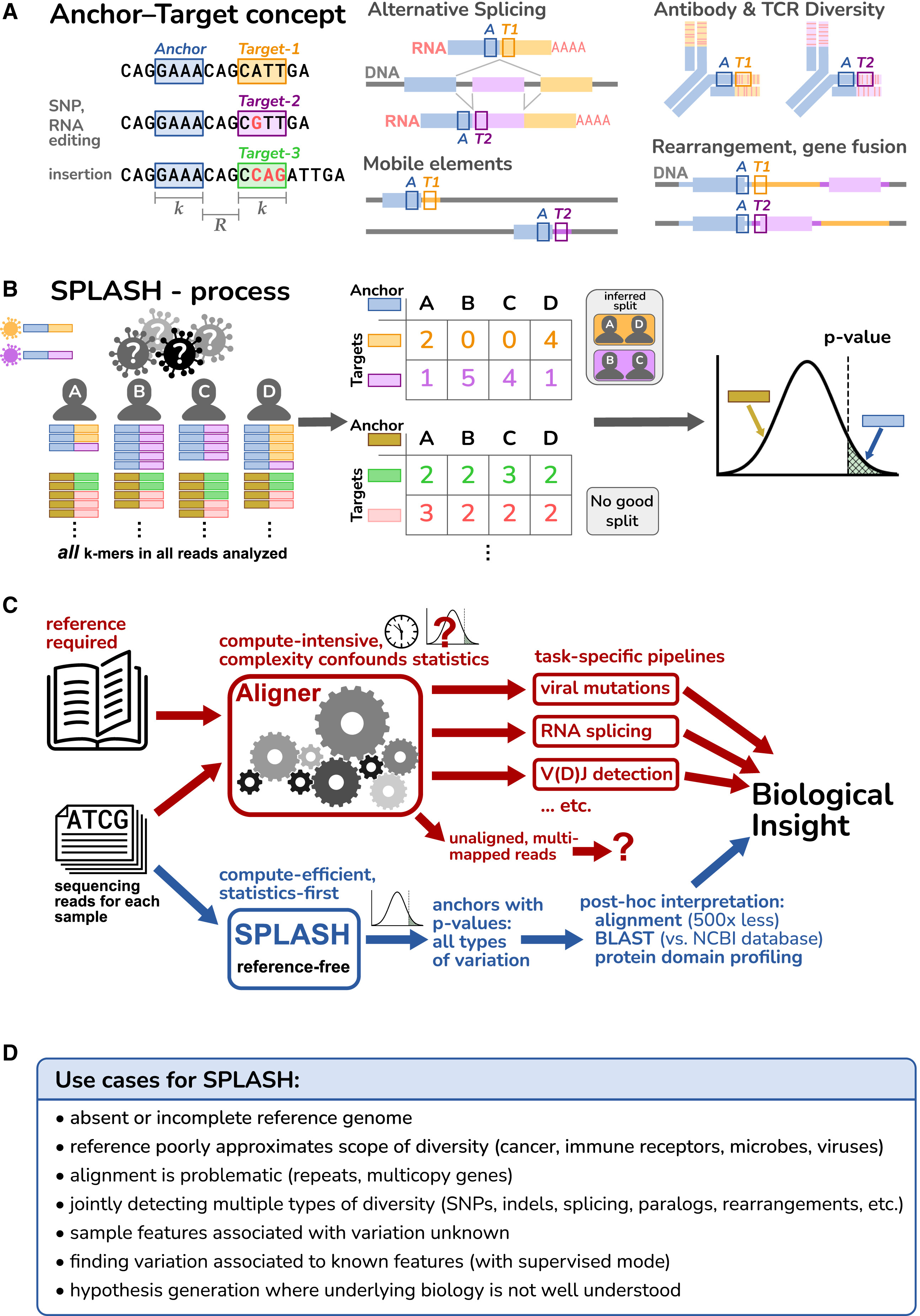
SPLASH概述
综上所述,SPLASH从“参考优先”方法转向“统计第一”,对原始测序数据进行统计假设测试。通过这种设计,SPLASH具有很高的计算效率。参考文献对解释很有价值;然而,通过参考对齐过滤数据会引入量化偏差和盲点。SPLASH承诺进行以前不可能进行的数据驱动的生物研究。
原始出处
Chaung, K., Baharav, T. Z., Henderson, G., Zheludev, I. N., Wang, P. L., & Salzman, J. (2023). SPLASH: a statistical, reference-free genomic algorithm unifies biological discovery. Cell, 186(25), 5440-5456.

