AJHG观点文章 | 展望新时代:来自常规T2T基因组的完整遗传信息
时间:2024-05-22 21:02:29 热度:37.1℃ 作者:网络
美国加州大学Karen H. Miga教授和华盛顿大学Evan E. Eichler教授合作在The American Journal of Human Genetics发表了观点文章“Envisioning a new era: Complete genetic information from routine, telomere-to-telomere genomes”,阐述了现阶段人类T2T基因组组装为人类遗传学领域带来的改变,预测了完整基因组时代对人类健康与疾病的多方面影响[1]。

背 景
人类端粒到端粒(T2T)染色体的完全测序代表了人类遗传学领域的一个关键变化。因为在原则上,所有形式的变异都可以被发现,而不分类别、频率或位置。到目前为止,对于基因组中的高度重复和结构复杂的区域来说,生成参考基因组仍存在技术限制。
解决这些问题的关键是长读长测序技术。长读长测序和组装的进步意味着人们可以生成比本世纪初“人类基因组计划” 的参考基因组更准确、更连续的定相基因组。虽然目前比短读长测序更昂贵,但随着长读长测序成本的下降和通量的增加,获得完整的T2T人类基因组测序预计将变得更加容易。这对我们如何映射、分类和将变异与人类疾病联系起来意味着什么?获得完整基因组测序将如何改变未来精准医疗的基础?我们怎样才能让更多的人受益于这项新兴技术呢?
作者认为,在未来十年中,长读长测序技术的技术进步可提供更为便捷的T2T定相基因组组装。世界各地的研究人员和临床医生对这些完整基因组信息的随时访问和自由决策将对扩大变异发现、了解遗传多样性和拓宽表观遗传特征产生影响。T2T基因组完全捕捉人类遗传变异图谱的潜力正开始改变人类遗传学领域,包括我们对遗传变异及其功能影响的理解,以及我们如何表示这种变异并将其与人类疾病联系起来。
01 获得完整基因组将改善与人类健康和疾病的遗传关联
人类遗传学的重点是建立基因型和表型之间的联系。而基因变异的发现依赖于技术的进步。虽然细胞遗传学、微阵列、光学图谱和短读长测序都能检测不同类型的遗传变异,但就其所能发现的变异类型、类别或大小而言,没有一种方法是全面的。这导致了对人类遗传变异和多样性的有限理解,并影响了对孟德尔遗传病和复杂遗传病的分析。
长读长测序技术,如Oxford Nanopore Technologies(ONT)的纳米孔测序技术和Pacific Biosciences(PacBio)的单分子实时(SMRT)测序技术,正在改变人类遗传学领域。首先,将检测结构变异(SV)的灵敏度提高了2-3倍。SV作为全基因组关联研究信号的可能性是SNV的3倍,影响基因表达的可能性是SNV的50倍。这意味着,长读长测序可不成比例地揭示影响更大的变异。其次,ONT和PacBio都涉及天然DNA测序,其中CpG甲基化可以很容易地区分。这使得个体和组织类型之间的表观遗传差异很容易被识别,从而发现将遗传变异与DNA修饰联系起来的新疾病机制。最后,当前长读长测序的长度和准确性意味着,现在可以产生定相的基因组装配,其中母亲和父亲的单倍型几乎是T2T的解析。此外,现在可以在没有父母数据的情况下,通过评估来自阶段性基因组组装内印迹基因位点的长读长测序数据的甲基化状态来实现这一点。这使我们不再认为人类基因组是3GBP,而是6GBP,其中所有变异都是完全序列解析的,并且相对于该单倍型上的所有其他遗传差异是定相的.
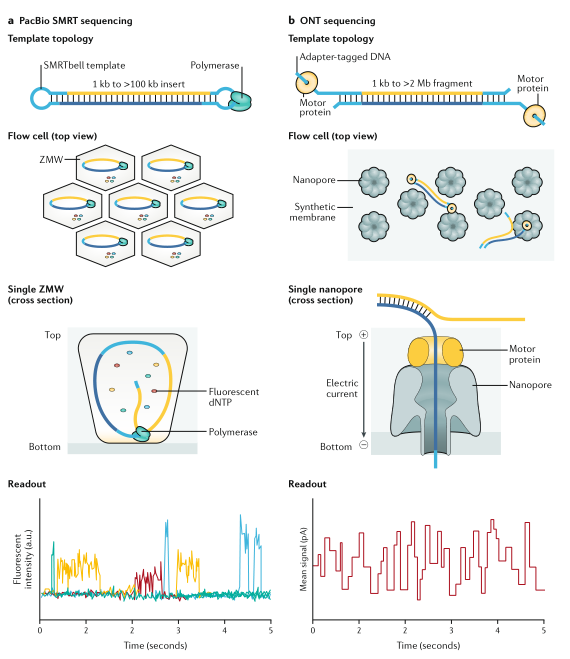
图1.长读长测序技术。来源:Nature Reviews Genetics[2]
原则上,不再需要遗传插补来重建单倍型,以前无法访问的基因组区域现在可以解析并访问遗传和表观遗传变异(图3)。这包括复杂区域的序列解析,例如SMN1和SMN2(图3b),它们是与脊髓性肌萎缩症(SMA)相关的基因治疗的重要靶点;完全解析脆性X综合征基因FMR1中的 CGG重复扩增,以及该重复序列的甲基化状态。总的来说,完全解析基因组的能力将为缺失的遗传率和新突变模型提供新见解,并建立与人类健康和疾病全面遗传关联的新标准。
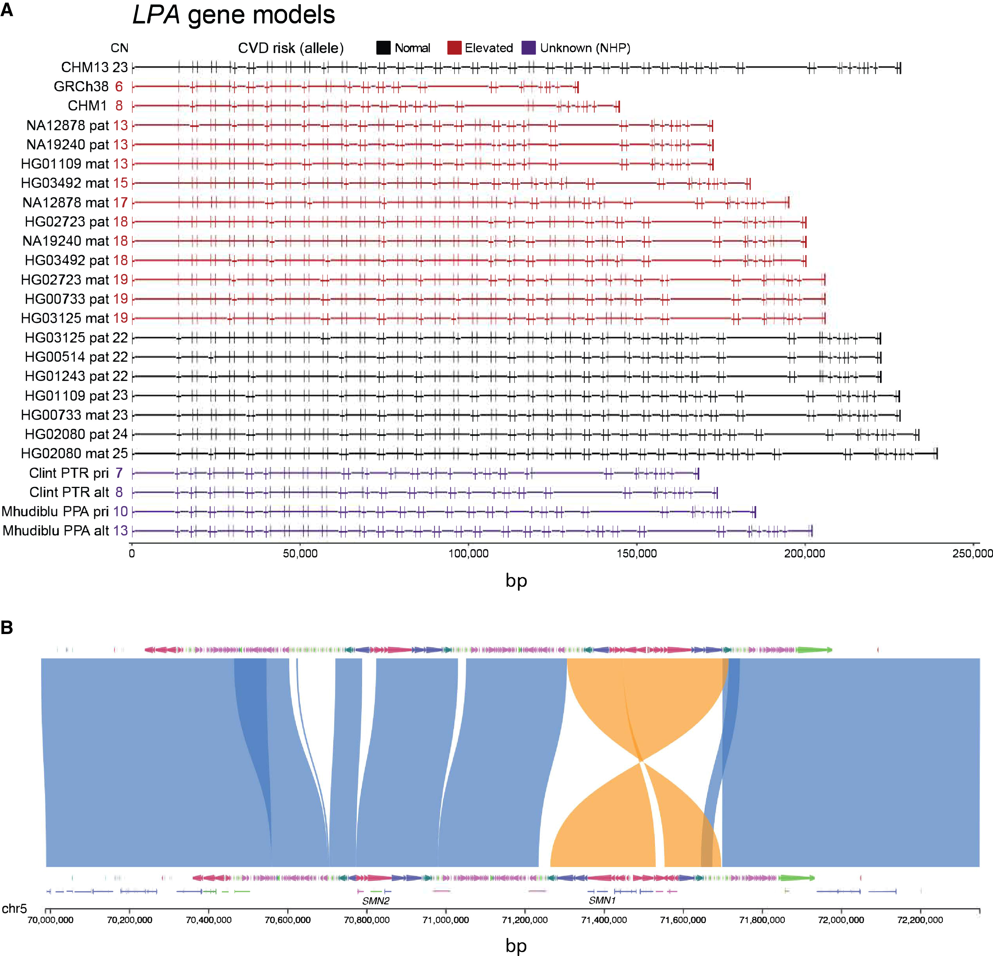
图3. 复杂的、生物医学相关的人类遗传变异的序列解析。来源:AJHG[1]
02 常规获取完整基因组需要技术的又一次变革
常规创建和分析T2T阶段基因组的成本、通量和计算基础设施目前仍然受到限制。在过去几年中,这一问题在很大程度上已经得到了解决——更精确的长读长测序和超长读长(>100 kbp)的结合已经引领了新组装算法的发展,如果能够产生足够的高分子量DNA,可以定相组装大部分人类基因组。T2T基因组的最佳实践是否会继续涉及PacBio和ONT平台的使用,还有待观察。但很明显,成本的下降和通量的增加,使定相基因组成为基础研究的一种可能。
除此之外,人类遗传学家面临的更大挑战是准确地描述发现的人类遗传变异的复杂性。与大多数人类遗传变异(即SNV)不同,这些变异涉及单个碱基从一个核苷酸到另一个核苷酸的变化,而许多新发现的变异在本质上更复杂,涉及结构变化、整个基因的拷贝数变化、位点间基因转换,甚至是基因组大部分片段(例如着丝粒、端着丝粒短臂)的完全进化更新。虽然过去的几年中开发了许多工具来可视化和理解这些区域的进化动力学,但对人类单倍型之间的这些差异进行分类并以标准VCF的形式记录它们是一项特别的挑战。然而,将这种变异置于人类单倍型的背景下是一个巨大的优势,使得更复杂基因分型工具的出现成为可能,以促进基因组复杂区域的基因分型。随着研究从一个基因组转向数百个基因组,很明显,一个参考基因组对于比较和理解人类变异的复杂性来说是不完全足够的基线。
03 全基因组参考图谱将代表常见和共享的单倍型
作者认为,有必要使人类参考基因组现代化,以更好地反映全球范围的基因组多样性。这一新的参考图谱将包含全球基因组更准确和多样化的变异,改善跨人群的疾病相关研究,将基因组学研究的范围扩大到基因组中最重复和多态的区域,并作为未来生物医学研究和精准医学的最终遗传资源。个体之间的遗传变异会影响疾病治疗的有效性,并影响患某些疾病的风险。通过创建更多样化的基因组参考,我们可以更好地了解这些变异,并开发出更个性化和更有效的医疗方法。此外,人类基因组的多样性反映了人类迁移和进化的复杂历史。通过在参考图谱中包含更多样化的基因组,我们可以更全面地了解人类的历史和进化。为此,人类泛基因组参考联盟旨在开发一种新的人类基因组参考图谱,称为“泛基因组”。这一新的泛基因组参考图谱将为识别和预测不同种群中变异的功能结果奠定重要基础。
新人类泛基因组参考的开发分为两个阶段。第一阶段为生成一系列几乎完整的单倍型解析基因组,其中所有基因和遗传变异都以线性方式表示,并且人类遗传变异的多样性得到充分调查。第二阶段为开发代表这种多样性的方法,其中共享部分与可变部分区分开来(图4),因此,所有后续人类基因组都可以比单个参考基因组更好地映射。新人类泛基因组参考的目标很简单:消除因映射到单个参考而引入的参考偏差,从而提高与该单个参考关系较远的基因组的映射和基因分型准确性。在计算基因组学领域,一个特别流行的方法是尝试开发泛基因组的图形模型。例如CYP2D6/D7基因位点,使用三种不同的方法构建了第一个使用47个人类基因组(94个单倍型)的人类全基因组图谱,从而更好地表征结构更复杂基因位点的变异。结果表明,泛基因组方法不仅在SV表征方面优于所有其他方法,而且在较小的变异(包括SNP和Indels)表征方面也优于所有其他方法,显著减少了错误并提高了全基因组的准确性。因此,人类基因组的常规生成是一项必要的努力,因为额外的基因组数据增强了泛基因组的稳健性和精确性,确保其仍然是映射、基因分型和解析人类遗传变异的最有效工具。
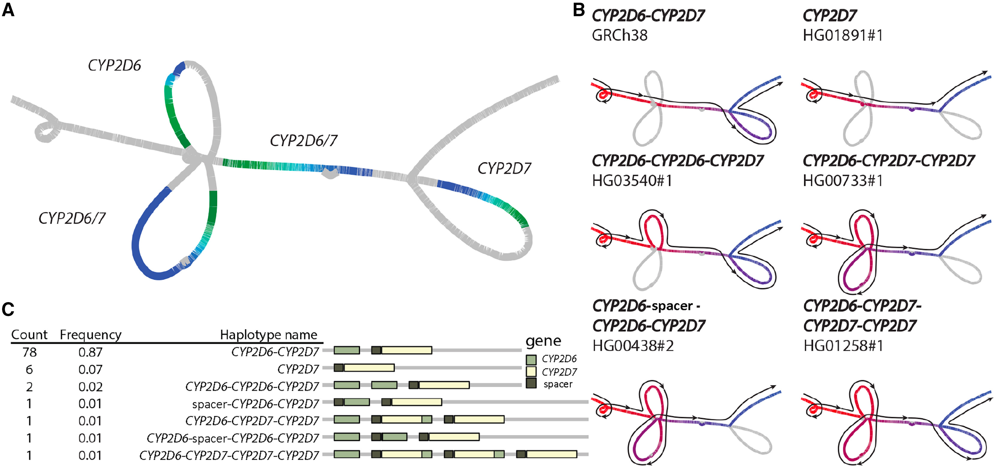
图4. 基于图表的CYP2D6/D7基因位点SV表示。来源:AJHG[1]
04 预测
作者认为完整遗传信息的附加值将使个体T2T基因组成为基础和临床研究的必然,并鉴于此,就其对各领域的影响做出五个大胆的预测。
1 了解新的突变机制及其与人类健康的关系
常规和全面的人类基因组测序和映射将彻底改变我们对突变机制及其对人类健康影响的理解。涉及的三种机制分别是:近端着丝粒DNA的异位交换,驱动着丝粒DNA卫星进化的重组过程和片段复制的位点间基因转化。例如,已有研究表明,在细胞分裂过程中,跨越着丝粒组装和着丝粒功能已知区域的卫星阵列结构的变化会影响染色体分离,因此对着丝粒卫星阵列突变机制的深入研究可为非整倍体研究提供了新视角,非整倍体与各种疾病如唐氏综合征、爱德华兹综合征和某些癌症有关。随着向常规的、完整的基因组图谱迈进,我们检测、研究和理解这些机制的能力将大大提高,为更有效的基因诊断和理解疾病易感性铺平道路。
2 重建人类基因组每个碱基对的进化关系
通过常规获取整个系统发育树中不同物种的综合基因组,我们可以完整测序人类密切相关物种(例如黑猩猩、大猩猩)的基因组,并重建人类基因组中每个碱基对的祖先状态。通过比较这些完整的基因组,我们可以分离出人类固定的遗传差异,并描绘出使人类作为一个独特物种的进化途径。揭示这些人类特有的基因变异以及其他灵长类动物的正常变异模式,可能会为人类健康和疾病提供前所未有的见解。有了T2T基因组,我们将能够在人群抽样中扩展我们的序列分类,并在重复序列丰富的复杂区域中表示新的SV。
3 单细胞基因组学扩大体细胞突变和细胞功能的范围
体细胞突变在人类健康和疾病中起着至关重要的作用,包括衰老和癌症。在生命早期生成个体的特异性T2T基因组组装的能力,将用于绘制所有随着个体年龄的增长而出现或克隆扩增的后续体细胞变化。个体的组装可以用于确定新的体细胞突变、表达变化以及甲基化差异。随着单细胞RNA和基因组测序从短读长到长读长的发展,检测这些基因变化的精度和准确性将变得非常高。值得注意的是,单细胞DNA扩增方面的重大技术进步首先需要从长读长测序中获益,因为目前的方案需要高分子量DNA,而这很难从单细胞中获得。
同时,快速确定单个细胞的完整遗传和表观遗传图谱的能力将更广泛地为细胞生物学和功能提供宝贵的见解,使我们能够绘制每个细胞的生命史,了解其当前状态并预测其未来行为。这种详细的图谱能以新维度揭示生物复杂性,指导对癌症等疾病的精确干预,并有可能在细胞水平上阐明衰老机制。因此,这种技术是推进个性化医疗以及对人类健康全面了解的一个有潜力的前沿。
4 个体的参考基因组将作为未来医疗记录的宝贵资源
常规获取受影响个体的全基因组测序将显著改善临床护理。每个人的基因组信息都可以作为个性化医疗的有力工具,使临床医生能够了解个体独特的基因组成和对疾病的潜在易感性,包括由于体细胞突变随时间推移而发生的变化。从本质上讲,受影响个体的基因组是其医疗记录的重要组成部分,影响着从疾病风险评估、早期检测到治疗干预的一切,有助于实现更准确的诊断,更有针对性的治疗,甚至是针对个人特定基因谱的预防策略。
5 扩大全球基因组学社区和信息共享,推进医疗保健
虽然基因组序列已被公开且不受限制地共享,但在谁能立即从获得人类参考基因组中获益——无论是在经济上还是在新药开发方面——仍然存在不平衡。因此,在强调实现T2T基因组常规获取的目标时,重要的是要认识到当前的这种不平等,并考虑在全球范围内分享利益。作者预测,基因组技术将有助于扩大常规基因组测序和分析的国际访问。这意味着长读长测序技术必须在全球范围内可负担并易于获得,相关测序仪器、实验试剂和耗材应进行优化,使其能够在不同环境温度下远距离运输。此外,新技术应该努力消除对昂贵实验室基础设施的要求,减少对高速互联网和数据处理计算资源的需求,并减少试验方案中对冰或干冰的需求。
结 语
我们正在进入一个全基因组信息将成为精准医疗和临床护理的重要驱动力的时代。因此,我们需要确保使用一项全球通用的技术来读取基因组,并且可供所有人使用。文中的大胆预测并不是纯粹的科幻小说,因为我们已经看到了早期的迹象,即在新冠疫情期间,全球团结起来使用测序技术追踪COVID-19变异来抗击新冠肺炎。我们相信,现在是共同努力确保这项新技术在全球范围内可用的时候了,这将推进精准医疗的发展,并改善人们的健康状况。
参考资料:
[1] Miga, K. H., & Eichler, E. E. (2023). Envisioning a new era: Complete genetic information from routine, telomere-to-telomere genomes. The American Journal of Human Genetics, 110(11), 1832-1840.https://www.cell.com/ajhg/fulltext/S0002-9297(23)00324-5
[2]. Logsdon, G. A., Vollger, M. R., & Eichler, E. E. (2020). Long-read human genome sequencing and its applications. Nature Reviews Genetics, 21(10), 597-614.

