盘点免疫检查点的配体
时间:2024-09-19 23:01:38 热度:37.1℃ 作者:网络
前言
在肿瘤免疫治疗中,免疫系统主要诱导细胞毒性淋巴细胞的细胞溶解作用来抑制疾病的进展。然而,肿瘤微环境中的CD8+T细胞往往因为持续的抗原刺激,发生耗竭。耗竭的CD8+T细胞持续存在,但逐渐失去其效应器功能、细胞毒性和增殖能力,导致免疫监视功能低下。
耗尽的CD8 T细胞通常表达一整套抑制性免疫检查点受体(ICR),由同源配体触发,通过下游信号通路调节T细胞反应。在耗竭的CD8+T细胞中,阻断ICR和免疫检查点配体(ICL)之间的相互作用是恢复CD8+T细胞的可行策略。考虑到肿瘤微环境中T细胞与其他免疫细胞之间的广泛相互作用,免疫细胞上的免疫检查点配体表达在促进抗肿瘤免疫应答方面与肿瘤细胞上的ICL表达同样重要。因此,有必要深入了解这些ICL,以及哪些因素导致ICL表达的患者间差异。
PD-1的配体
PD-1介导的T细胞抑制可归因于PD-1的两种众所周知的配体,即PD-L1和PD-L2(B7-DC、CD273)。
PD-L1
相比PD-L2,PD-L1是PD-1更主要的配体。肿瘤细胞的PD-L1表达通常发生在恶性转化过程中,然而,除了导致PD-L1在肿瘤细胞中组成性表达的内在因素外,肿瘤微环境中的外在因素也可促进PD-L1的表达,如营养缺乏、代谢物积累和缺氧。
炎症性肿瘤微环境提供了调节PD-L1表达的各种因子。IFN-γ主要由效应T细胞和NK细胞分泌,是各种肿瘤细胞PD-L1最有效的诱导剂。在机制上,IFN-γ诱导的PD-L1上调是通过JAK1/2–STAT1激活介导的,最终由干扰素调节因子1(IRF1)与PD-L1启动子直接结合。除干扰素外,其他炎症介质也调节PD-L1的表达,如TNF-α、IL-6、IL-27、TGFβ等。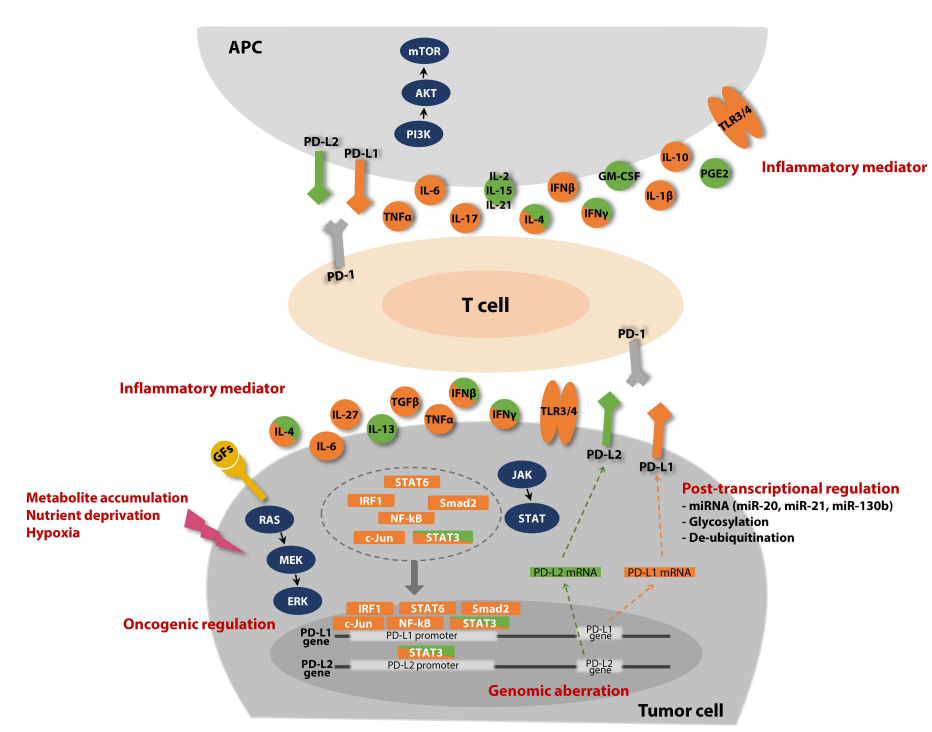
肿瘤浸润性免疫细胞表达的PD-L1通过多种机制促进免疫逃逸。在抗原呈递细胞(APC)上表达的PD-L1,包括树突状细胞和巨噬细胞,与表达PD-1的T细胞或共刺激分子CD80顺式相互作用传递抑制信号。此外,来自活化T细胞的PD-L1与其他T细胞或巨噬细胞上表达的PD-1结合,后者促进M2极化。T细胞上表达的PD-L1本身作为受体,可导致活化T细胞的无能状态或凋亡。
PD-L2
作为PD-1的第二配体,PD-L2也赋予PD-1抑制功能,尽管其机制尚不完全清楚。PD-L2以比PD-L1更高的亲和力与PD-1结合,PD-L2的表达最初被认为仅限于树突状细胞或巨噬细胞,但最近的研究表明PD-L2的表达没有以前认为的那么受限制,已有报道表明其表达在许多人类恶性肿瘤中。
与PD-L1一样,PD-L2在肿瘤细胞中通过IFN-γ诱导上调,IFN-β通过促进STAT3与PD-L2启动子的相互作用也具有类似的作用。此外据报道,Th2型细胞因子IL-4和IL-13也可诱导食管腺癌中PD-L2的表达。
在免疫细胞中,IFN-γ和Th2细胞因子参与PD-L2的表达。粒细胞-巨噬细胞集落刺激因子(GM-CSF)是巨噬细胞和树突状细胞中PD-L2的诱导剂。此外,常见的γ链细胞因子,如IL-2、IL-15和IL-21,可在单核细胞或巨噬细胞中诱导PD-L2表达。
TIGIT的配体
TIGIT是一种抑制性受体,主要由NK细胞、调节性T细胞、记忆性T细胞和耗竭的CD8 +T细胞表达。人TIGIT可与三种配体结合,即脊髓灰质炎病毒受体(PVR,NECL5,CD155)、PVR相关2(PVRL2,NECIN2,CD112)和PVR相关3(PVRL3,NECIN3,CD113),其中PVR对TIGIT的亲和力最高。PVR/TIGIT结合通过磷酸化TIGIT胞内段的免疫受体酪氨酸基抑制基序(ITIM)或干扰PVR/DNAX相关分子1(DNAM-1,CD226)结合抑制T细胞反应。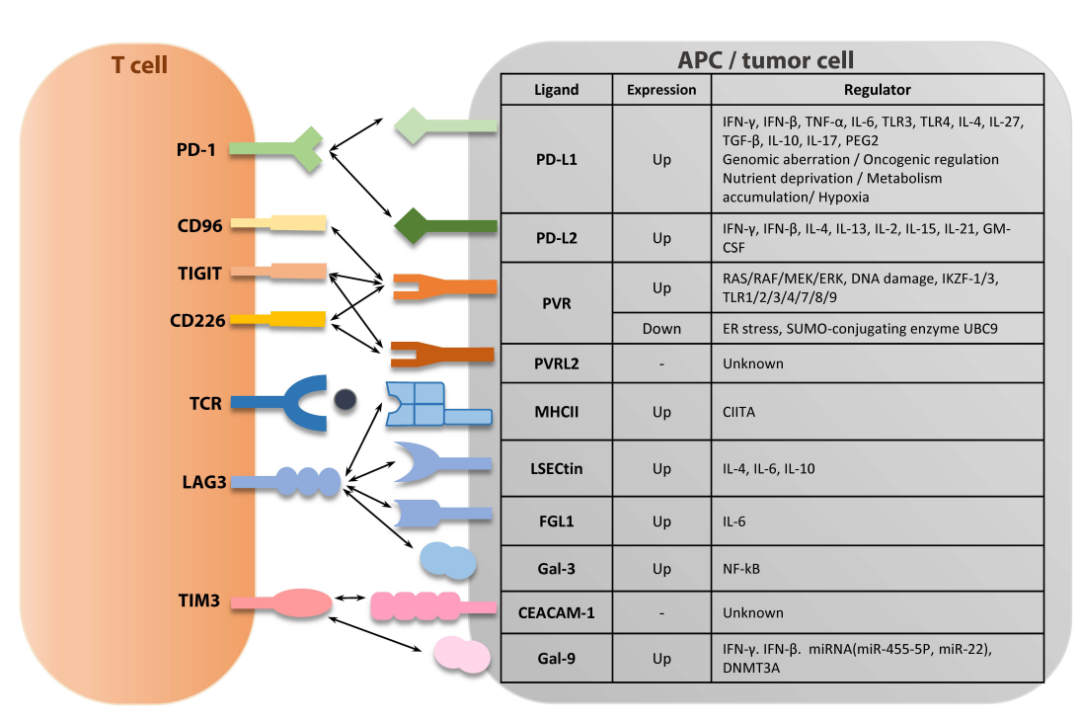
PVR是一种粘附分子,在许多类型的实体和血液系统恶性肿瘤中过度表达,其过度表达与不良预后相关。PVR是连接蛋白和连接蛋白样(Necl)分子家族的成员,并参与各种生理过程,包括细胞间粘附、运动、增殖和分化,其过度表达可能以肿瘤细胞固有的方式产生促肿瘤作用。同时,致癌的RAS/RAF/MEK/ERK信号通路通过激活蛋白-1(AP-1)与小鼠成纤维细胞中PVR启动子的直接结合来上调PVR的表达,但尚不清楚RAS/RAF/MEK/ERK信号是否也增加了人类肿瘤细胞中PVR的表达。
然而,就抗肿瘤免疫反应而言,PVR过度表达是否具有促肿瘤或抗肿瘤作用仍不确定。在调节抗肿瘤免疫反应时,PVR可与三种不同的受体结合——DNAM-1、TIGIT和CD96,根据其结合的受体,可能会产生矛盾的效应。许多研究表明PVR是NK细胞功能的刺激因子,然而,鉴于TIGIT与PVR的结合亲和力高于DNAM-1,且TIGIT在肿瘤浸润淋巴细胞上高度表达,因此,肿瘤细胞上的PVR表达如何影响体内效应细胞需要进一步研究。
TIM-3的配体
TIM-3是T细胞耗竭的标志,是免疫治疗中最常见的靶点之一。Tim-3有四种配体:半乳糖凝集素-9(Gal-9)、磷脂酰丝氨酸(PtdSer)、高迁移率族蛋白盒1(HMGB1)和癌胚抗原相关细胞粘附分子1(CEACAM1)。
其中,Gal-9和CEACAM1通过从TIM-3分离HLAB相关转录物3(BAT3)来减弱TCR信号,并最终导致细胞凋亡。此外,TIM-3和CEACAM1之间的细胞内相互作用支持TIM-3的成熟和表面转运。因此,T细胞内缺乏CEACAM1会导致TIM-3在细胞内积聚,并且无法与配体相互作用,从而使T细胞从TIM-3介导的抑制中释放出来。
另一方面,暴露在凋亡细胞外小叶上的PtdSer可触发CD8+树突状细胞或巨噬细胞亚群上表达的TIM-3并诱导吞噬作用。而HMGB1,一种报警蛋白,可以释放到肿瘤微环境中,与游离DNA形成复合物,复合物的形成有助于DNA内化到树突状细胞中以激活内体TLR。而TIM-3结合HMGB1可抑制复合物的形成和随后的树突状细胞激活。迄今为止,已知TIM-3与PtdSer或HMGB1之间的相互作用间接影响T细胞功能,但PtdSer或HMGB1是否直接影响表达TIM-3的T细胞还有待研究。
LAG-3的配体
迄今已发现了五种LAG-3配体:MHC II、半乳糖凝集素-3(Gal-3)、肝窦内皮细胞凝集素(LSECtin)、α-突触核蛋白原纤维(α-syn)和纤维蛋白原样蛋白1(FGL1)。与其他配体不同,LAG-3结合α-syn与神经系统中病理性α-syn纤维的细胞间传递有关,与免疫反应无关。
尽管有报道称,LAG-3可以高亲和力结合MHCII,并通过阻止MHCII和CD4之间的相互作用来调节CD4+T细胞,但最近的研究表明,LAG-3与MHCII/肽复合物(pMHCII)的结合通过转导抑制信号抑制CD4+T细胞。
此外,研究还发现,大量表达pMHCII的APC通过LAG-3依赖机制抑制CD8+T细胞的激活。另外,其他配体包括Gal-3、LSECtin和FGL1与LAG-3结合,都可以对肿瘤微环境中的CD8+T细胞进行负性调节。
肿瘤内CD8+T细胞和基质细胞是Gal-3的主要来源,但Gal-3也可由多种类型的肿瘤细胞分泌。LSECtin或FGL1在正常生理条件下在肝脏中表达,在一些肿瘤细胞中高度上调。在肿瘤细胞中通过IL-6或IL-10可诱导LSECtin表达,或通过IL-4也可以在人类单核细胞来源的树突状细胞中诱导LSECtin表达。此外,L-6还增加人肝癌细胞中的FGL1的表达。未来的研究需要验证这些配体的精确分子机制以及它们在LAG-3介导的T细胞抑制中的作用。
VISTA的配体
对于VISTA配体的研究长期处于空白状态。然而,目前多个已发表的研究都强调了潜在的VISTA配体。最近,一项研究确定VSIG3(或 IgSF11)为VISTA配体,这种相互作用有助于抑制T细胞反应。由于VSIG3在造血细胞中的表达是无法检测到的,这种相互作用在体内的生理相关性仍有待验证。
此外,有研究发现,VISTA和P-选择素糖蛋白配体-1(PSGL-1)之间存在pH依赖性相互作用。研究人员强调了VISTA细胞外结构域的一个独特特性,它富含组氨酸残基。组氨酸的侧链在酸性pH条件下发生脱质子化,例如在TME中发现的侧链,这种翻译后修饰允许与 PSGL-1 结合。鉴于VISTA多聚体(包括 VISTA-Fc)只在酸性条件下与白细胞结合,这种翻译后修饰在决定VISTA配体相互作用中的重要性是不可低估的。
对这一发现的热情有多种原因。首先,PSGL-1和VISTA在调节T细胞活性和肿瘤免疫方面的作用越来越受到重视。其次,pH 和离子环境是调节免疫受体相互作用的一个新的和有价值的概念。这在离子环境在明显影响T细胞免疫命运的肿瘤中尤为重要。更重要的是,这项研究提出了一种创新的策略来构建pH选择性抗体,即配体只针对pH失调的部位(如肿瘤)。
小结
在肿瘤微环境中,肿瘤细胞通过诱导表达和调控不同的ICR和ICL,建立了一个适宜生长的环境。随着肿瘤的发展,ICRs同时受到共同因素的诱导,而相应ICLs的表达具有重叠但独立的调控。此外,肿瘤微环境中存在多种调节因子,每种调节因子都可能对ICL表达产生不同的影响。
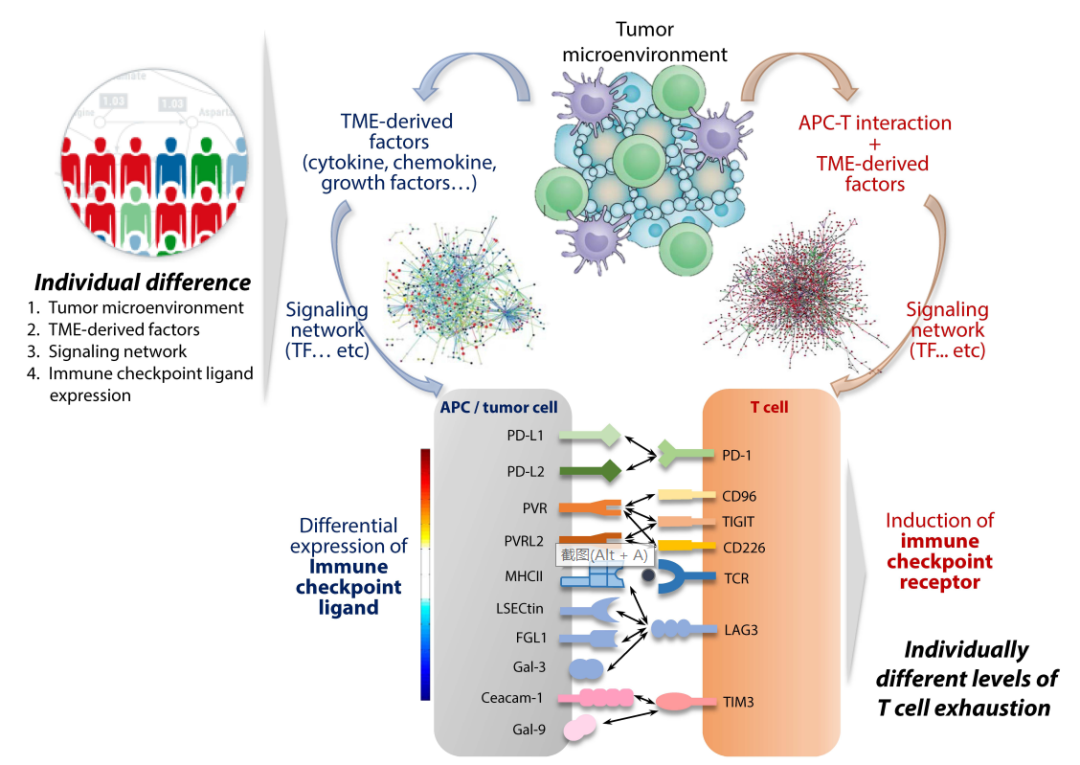
由于ICL表达模式可能指示免疫检查点途径的活动,个体间ICL的差异表达谱可用于预测免疫检查点阻断的治疗反应。例如,许多研究表明PD-L1表达与PD-1阻断剂的治疗反应相关。因此,通过识别基于ICL的稳健生物标记物,有可能实现更高的应答率和精确的免疫治疗。
参考文献:
1.Perspectives on immunecheckpoint ligands: expression, regulation, and clinical implications. BMB Rep. 2021Aug 31; 54(8): 403–412.
