Leukemia:RUNX1基因可能与儿童急性髓系白血病不良预后有关
时间:2023-07-23 13:27:42 热度:37.1℃ 作者:网络
急性髓系白血病(AML)约占儿童白血病20%,相比急性淋巴细胞白血病(ALL)预后更差,5年生存率约68%。近年来,对肿瘤驱动基因和预后之间关联的研究逐渐深入,根据驱动基因可将AML分成预后不同的风险组,而风险组别会采取差异化的治疗策略。然而,多数关于基因与预后关联的研究是在成人患者队列中开展的。儿童AML的分子生物学景观与成人不同,因此,有必要对儿童的AML的分子学特征及其对预后的影响展开研究。
RUNX1基因编码一个在造血干细胞中表达的转录因子,该转录因子参与造血干细胞的分化。既往研究发现,RUNX1基因的突变参与白血病的发生。在成人AML人群中,RUNX1突变在5-8%的较为年轻的患者人群中被发现,并且与M0 FAB亚型、正常核型和不良预后有关。由于RUNX1突变的患者具有不良预后,携带RUNX1突变的成人患者被归入高危组。相比RUNX1突变,RUNX1缺失的研究相对较少,因此其对预后的影响是相对未知的。对于儿童AML,由于RUNX1突变和缺失发生率较低,因此其对预后的影响未知,也没有被纳入危险分层和治疗决策中。
近日,Lucille Lew-Derivry 等研究人员在 Leukemia 上发表了题为 Prognostic impact of RUNX1 mutations and deletions in pediatric acute myeloid leukemia: results from the French ELAM02 study group 的论文,为阐明RUNX1基因的改变对儿童AML预后的影响,该研究利用法国ELAM02儿童AML队列的数据进行回顾性分析,显示存在RUNX1基因改变的AML患儿预后更差,需要考虑将其纳入风险分层之中。

ELAM02研究共纳入438名新发AML患儿,其中386名患儿有RUNX1基因突变情况的数据;386名患儿中,29名(8%)具有RUNX1基因的改变,其中24人为基因突变,5人为基因缺失。因为RUNX1基因突变多为功能丢失突变,所以将突变和缺失合并成一组,RUNX1功突变/缺失组,和RUNX1野生型组进行比较和分析。
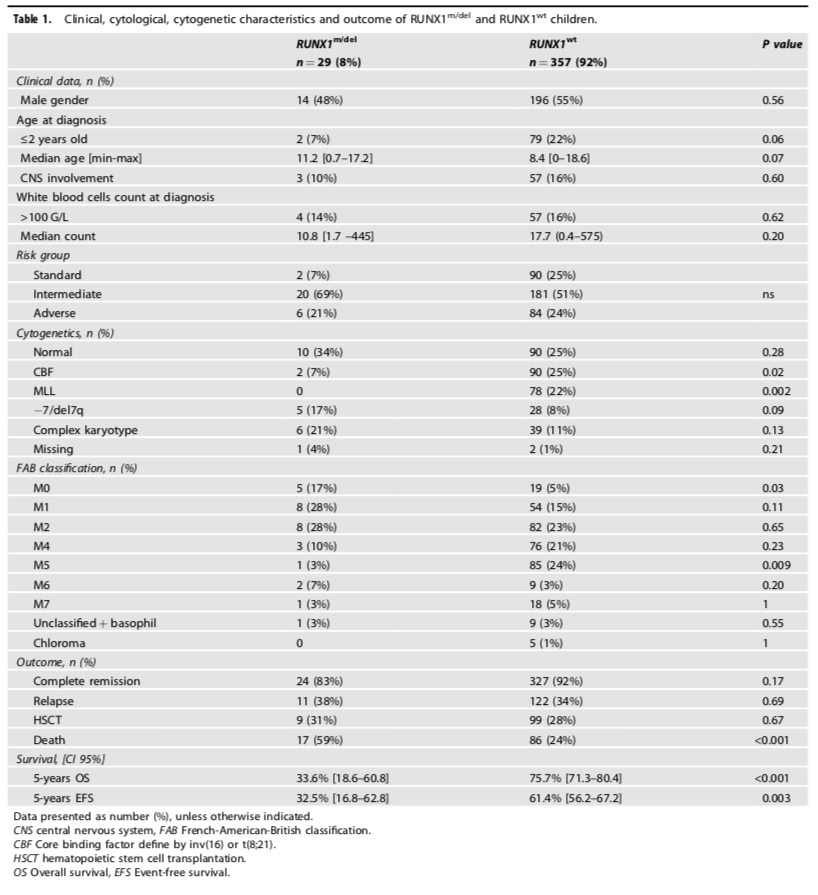
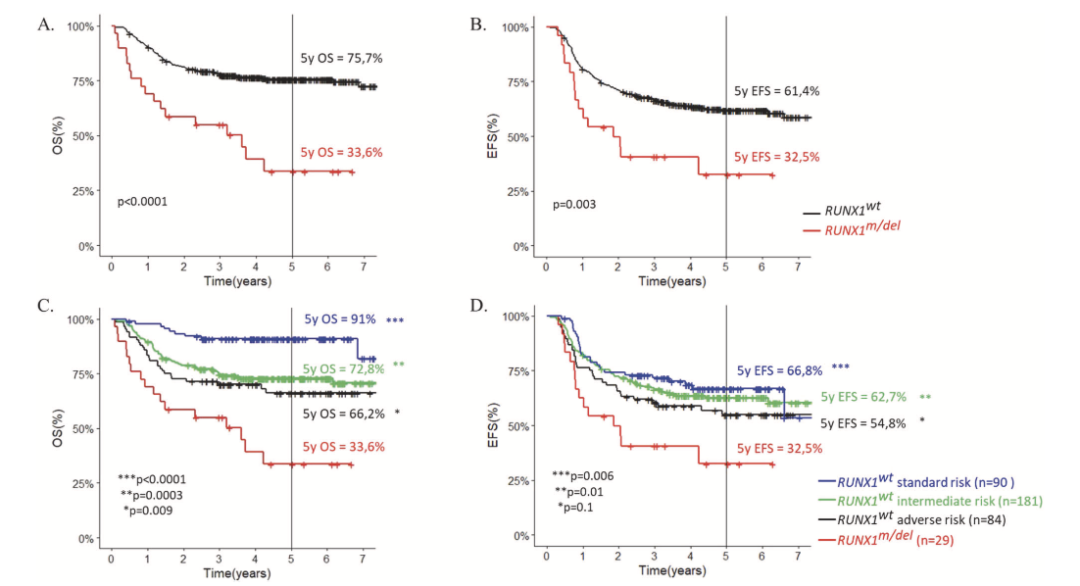
表1显示了RUNX1突变/缺失组和RUNX1野生型组的临床、形态学和遗传学特征。两组患儿在基线的临床特征上,如性别、年龄、白细胞数和中枢神经系统受累方面,没有统计学显著的差异。在形态学特征上,RUNX1突变/缺失组更多具有AML-FAB M0表型【5/29 (17%) vs 19/357 (5%), p = 0.03】,而不具有AML-FAB M5表型。细胞遗传学特征上,RUNX1突变组具有正常核型的比例更高【10/24 (42%) vs 90/357 (25%), p = 0.09】,并且与KMT2A (11q23)重排不同时出现,也罕见与CBF异常t(8;21)(q22;q22)同时出现。因此,RUNX1突变/缺失组中69%的患儿被归入中危组。
撰文

责编


