扫描隧道显微镜 | 抑制CCR7-PI3K信号可逆转ALK重排严重的负载问题
时间:2023-07-24 19:19:50 热度:37.1℃ 作者:网络
间变性休止休克(ALK)酪氨酸激酶抑制剂(TKI)在多种ALK驱动的肿瘤中常表现出良好的治疗响应,但其诱发性常导致患者休克。目前,ALK TKI的机制主要集中于ALK急性的非小细胞肺癌,而在ALK驱动间变性大细胞突变(ALK+ ALCL)中,ALK TKI的因果机制尚鹦鹉。
与非小细胞肺癌不同,在ALK驱动间变性大细胞肿瘤中,ALK TKI未能获得性的点突变,而是由旁路激活导致的,常见的信号故障有STAT3、MAPK等,提示ALK+ ALCL细胞在TKI压力下的长期可能与有利的血管周围肿瘤微环境有关。
近日,来自意大利的 Claudia Voena 团队和美国的 Roberto Chiarle 团队在 Science Translational Medicine 发表题为 Targeting CCR7-PI3Kγ overcomes resistance to tyrosine kinase inhibitors in ALK-rearranged lymphoma 的文章,对ALK TKI耐药的ALCL患者肿瘤组织和ALCL细胞系RNA测序结果分析发现,C-C基序趋化因子受体7(CCR7)可通过激活磷脂酰肌醇3激酶γ(PI3Kγ)信号维持有利于ALK+ ALCL细胞存活的肿瘤微环境,通过抑制PI3Kγ或阻断CCR7信号联合ALK抑制剂可降低ALK+ ALCL的原发耐药。

首先,为探索ALK TKI治疗失败的ALK+ ALCL牵连机制,研究团队对来自ALK TKI牵涉的ALK+ALCL儿童和ALCL细胞系的RNA高转录数据进行分析。在ALK+ALCL中,发现并确认克唑替尼干预与PI3Kγ信号表达上调有关。研究分析了ALK TKI牵涉的ALK+ALCL儿童患病RNA高通量干预数据,发现克唑替尼介入的患者和细胞中PI3Kγ表达上调,并且该结果在多种ALK+ ALCL细胞系中进行了验证。
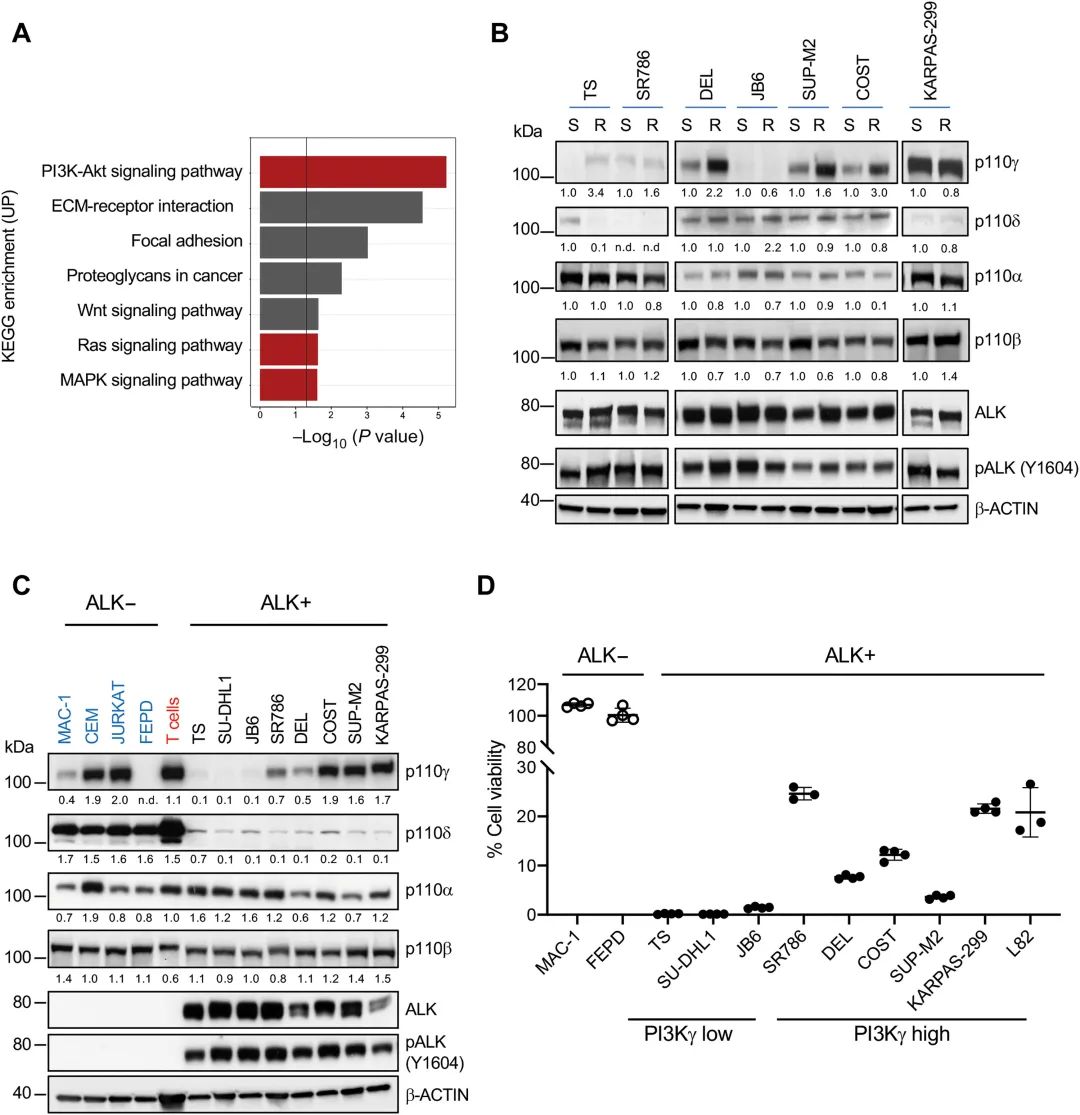
ALK+ALCL对克唑替尼的偶联性与PI3Kγ上调有关,该图(a)克唑替尼对的ALCL患者和细胞系中PI3K-Akt信号无法激活、(b)克唑替尼对的ALk+ALCL细胞系中PI3K的p110γ催化亚单位蛋白水平显着着升,(c)不同细胞系中PI3K包括体的表达情况,以及(d)PI3Kγ表达高的ALK+ALCL细胞对克唑替尼。
其次,研究人员发现PI3Kγ高表达可导致ALK+ALCL对克唑替尼。在PI3Kγ低表达的细胞系中外源性的过表达PI3Kγ或持续激活PI3Kγ,亟需诱导克唑替尼治疗组合;且通过分析对克唑替尼同时和长时间敏感的患者中PI3Kγ表达水平,发现PI3Kγ高表达的患者均对克唑替尼没有反应。
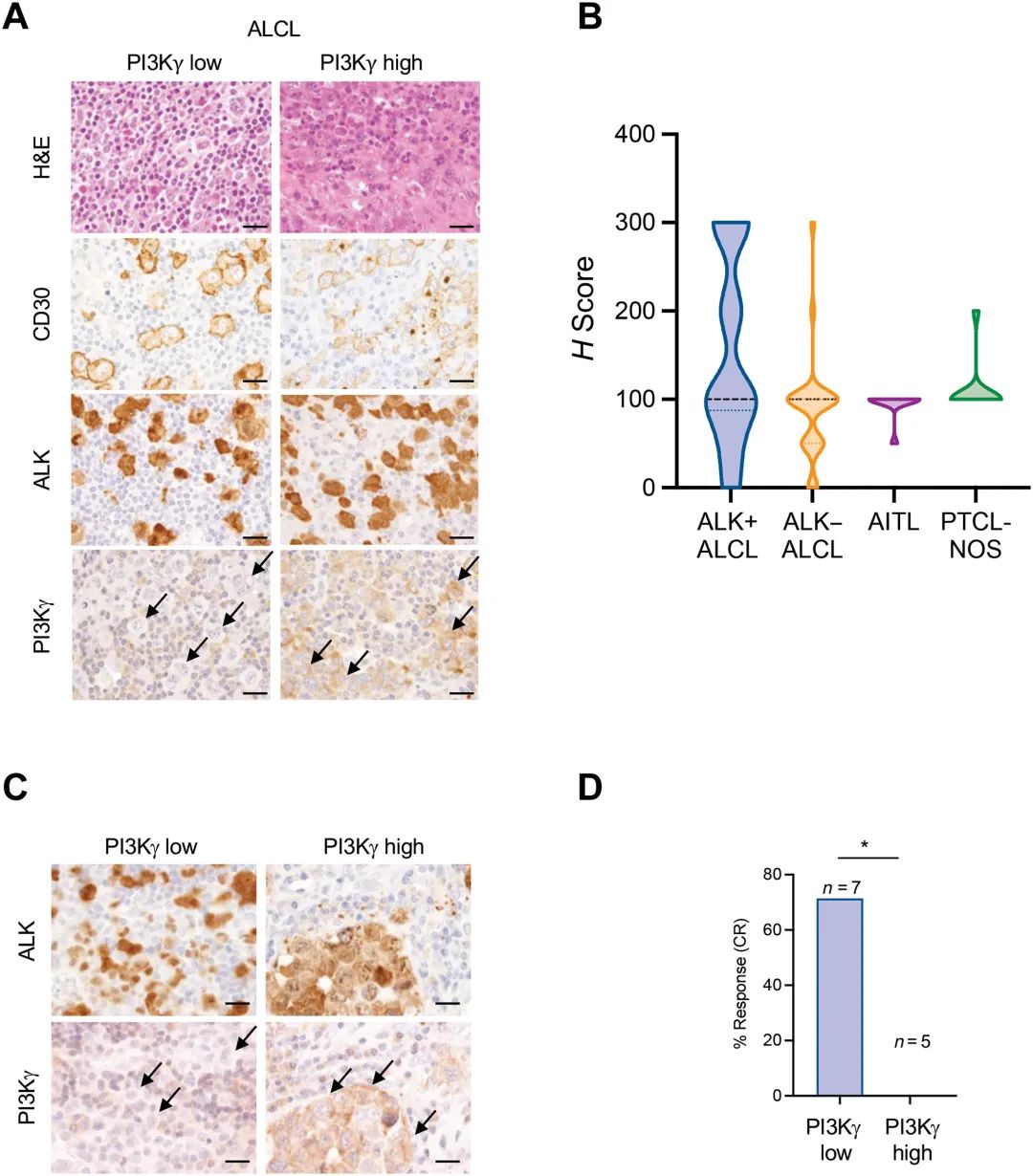
(a)PI3Kγ高表达和低表达ALCL患者的代表HE和免疫组化染色。(b)不同的对应亚型中PI3Kγ表达差异,(c)ALK+ ALCL患者中PI3Kγ和ALK免疫组化表达情况,以及(d)PI3Kγ高表达和低表达的ALK+ALCL患者对克唑替尼治疗响应的一半。
然后,联合度恩西布(duvelisib,PI3Kγ/δ染料)可显着增强克唑替尼抑制ALK+ALCL细胞生长作用。在PI3Kγ高表达的ALK+ALCL细胞系和PDXs模型中,单独使用度恩西布对细胞复制无影响,联合克唑替尼可显着增强其细胞增殖和细胞增殖作用。
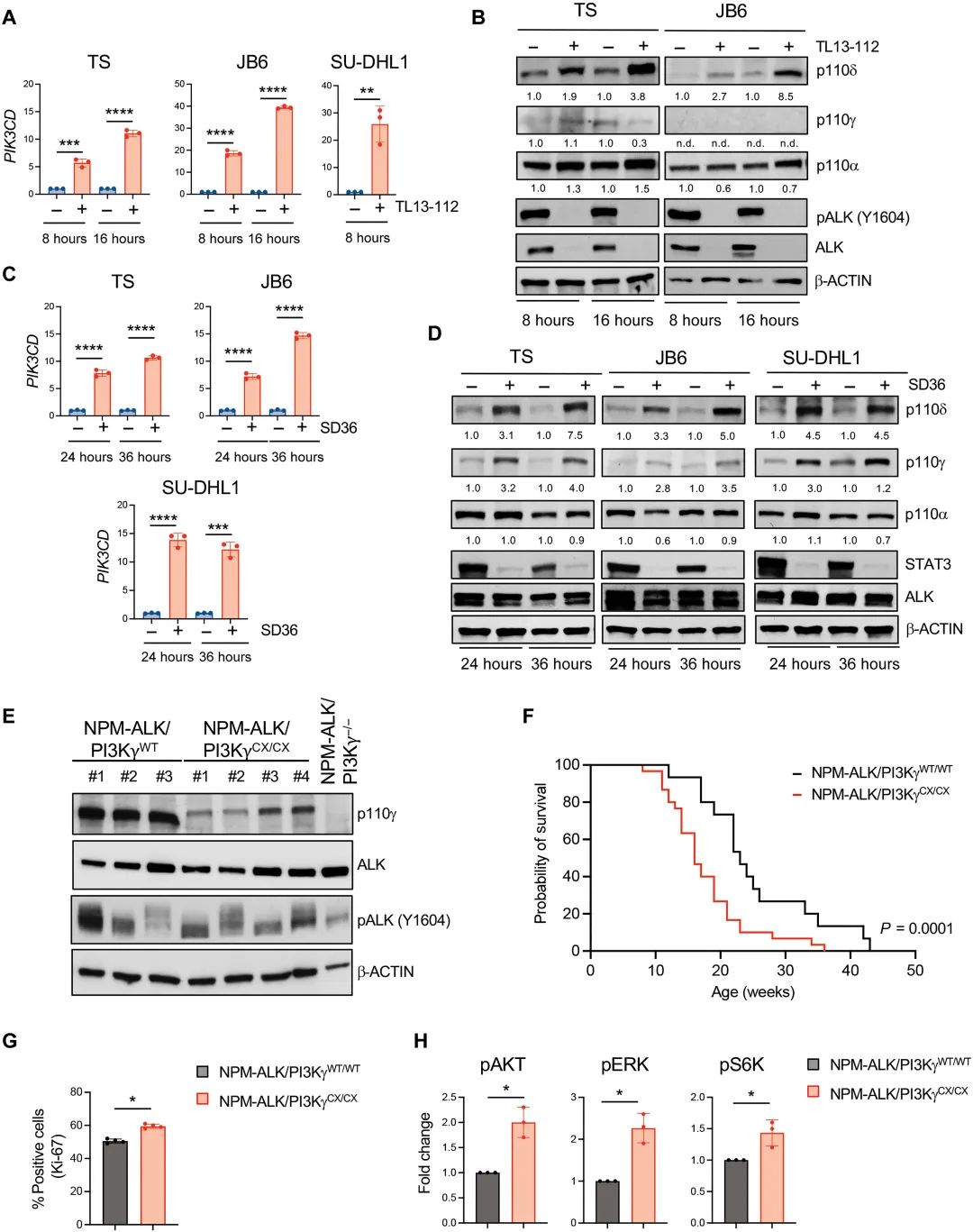
在ALK+ ALCL移植瘤模型中,度恩西布可增强克唑替尼治疗。(a)不同治疗处理ALK+ALCL异种移植瘤中,移植瘤Ki-67和Cleaved caspase-3免疫组化染色,(b)不同治疗处理ALK+ALCL异种移植瘤中Ki-67(上)和Cleaved Caspase-3(下)细胞的概念分析,(c和d)ALK+ ALCL和Cleaved caspase-3免疫组化染色,(c和d)ALK+ ALCL和Cleaved caspase-3免疫组化染色药物治疗探针以及体积变化情况,以及(d和e)接受不同治疗的小鼠移植瘤中ALK的表达和PI3K信号激活情况。
进一步的研究发现,在克唑替尼加入的ALK+ ALCL细胞中,CC基序趋化因子7(CCR7)的表达显着升高,且ALK+ALCL肿瘤周围的血管和间质细胞通过表达CCR7的体CCL19/21创造有利于ALK+ ALCL长期持续的微环境,抵抗克唑替尼诱导的诱发。
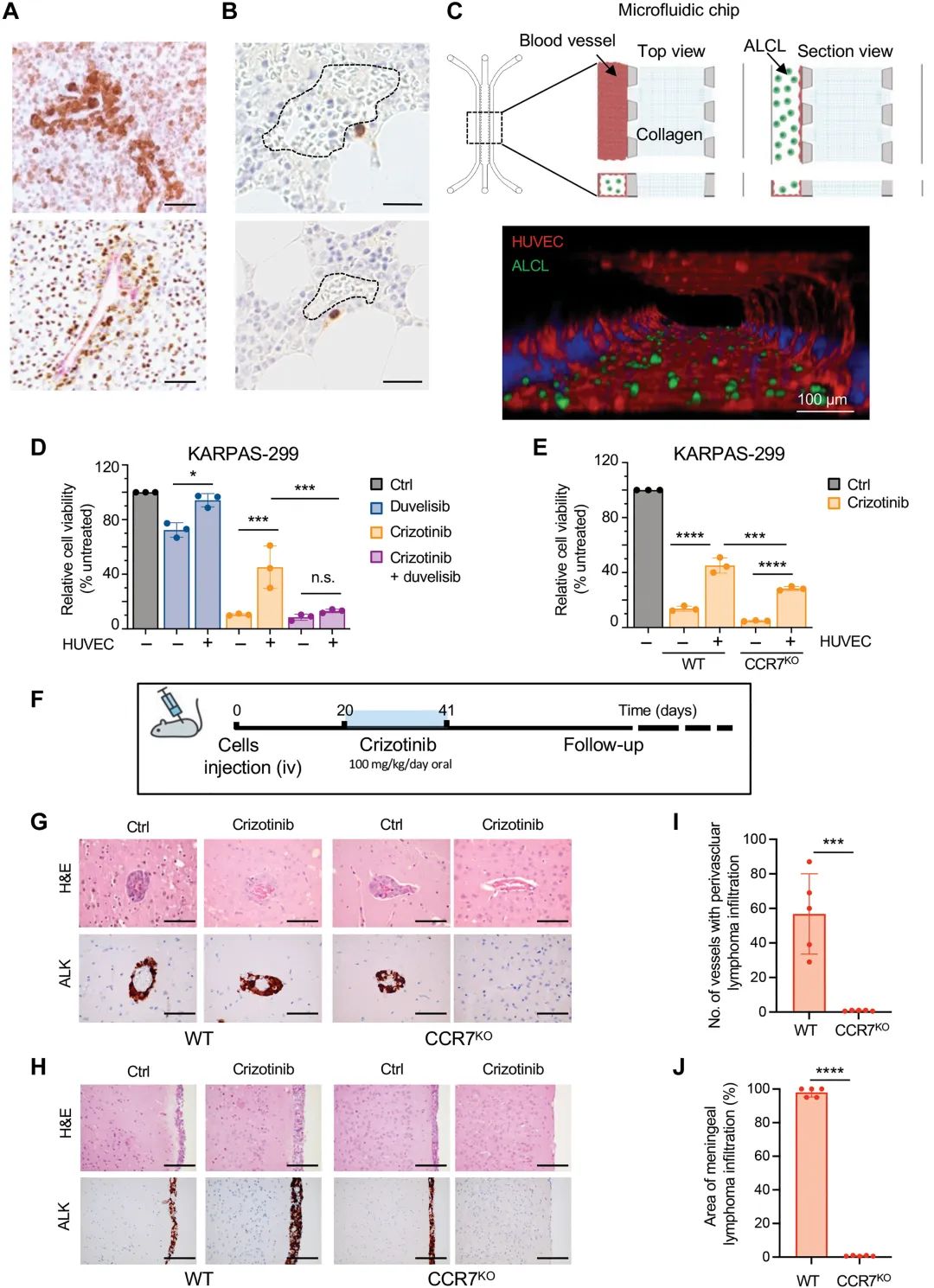
(a)原发性ALK+ ALCL染色中显示ALK+细胞在血管周围,(b)临床治疗的ALK+ ALCL患者骨髓中检测到在血管周围分布的罕见持久性ALK+细胞(稀有持续性ALK+细胞,棕色染色),(c)上图:微流控芯片示意图(左)和三维微流控模型中ALCL-肿瘤反应和血管形成的微物理模型示意图(右),底部:微流控模型中大血管(F-肌动蛋白,红色)和ALK+ ALCL(GFP,绿色)的共聚焦显微镜视野。(d和e)CCR野生型KARPAS-299(ALK+ ALCL细胞)和CCR7敲除KARPAS-299存在于HUVEC(人脐带血内皮细胞)细胞存在或不存在的情况下,克唑替尼的治疗反应存在显着差异,(和,g,h,i) j)在静脉注射COSTCCR7WT和COSTCCR7KO(ALK+ ALCL细胞)成肿瘤肿瘤模型上经克唑替尼治疗后,CCR7敲除模型中血管周(G)或脑膜(H)周围ALK+ALCL细胞,以及脑血管周围血管(I)或脑膜(J)肿瘤细胞都显着降低。
总之,该研究确定了ALK+ ALCL中酪氨酸激酶抑制剂(TKI)耐药的旁路机制,该机制由CCR7-PI3Kγ信号传导通路介导,在ALK TKIs治疗期间维持促细胞生存信号传导。文章指出PI3Kγ和CCR7可作为预测ALK +ALCL对ALK TKI疗效的潜在生物标志物;也可通过靶向CCR7/PI3Kγ信号增强ALK+ ALCL细胞对ALK TKIs疗效或克服其耐药,如联合度恩西布(PI3Kγ/δ双靶点抑制剂)和克唑替尼(ALK TKI);另外,本研究还发现可通过阻断CCR7联合ALK TKI预防或减少ALCL细胞在中枢神经系统的播散。
虽然本研究主要基于细胞和小鼠模型,但其发现的ALK+ALCL对ALK TKI的新的相互作用机制,为临床预测的生物标志物和预防的治疗思路:通过检测和阻断CCR 7/PI3Kγ来预测和遏制ALK+ ALCL对ALK TKI的相互作用。但将该诊断途径应用于临床实践中还需要进一步的临床试验验证。

