收藏 |成人和儿童中性粒细胞减少症的欧洲诊疗指南:EHA与EuNet-INNOCHRON COST行动共识
时间:2023-04-10 20:05:01 热度:37.1℃ 作者:网络
中性粒细胞减少症作为一种孤立的血细胞缺乏症,是各种获得性或先天性、良性或癌前疾病的特征,易发生骨髓增生异常肿瘤/急性髓性白血病,可发生于任何年龄。近年来诊断方法的进展,特别是基因组学领域的进展,揭示了负责病因学和疾病进化的新基因和机制,并为量身定制的治疗策略开辟了新的前景。尽管该领域的研究和诊断取得了进展,但来自国际中性粒细胞减少症患者登记和科学网络的真实证据表明,中性粒细胞减少症患者的诊断和管理主要基于医生的经验和当地实践。总体而言,血液科医生需要持续接受教育,了解已知和新出现的关于中性粒细胞减少症的诊断、随访和治疗策略尤为重要。
基于中性粒细胞减少症领域的研究进展及转化为临床实践、基因组学的进展和常规实践中基因检测的可用性,以及长期患者登记的累积经验,欧洲血液学协会 (EHA) 与欧洲慢性中性粒细胞减少症创新诊断和治疗网(European Network for the Innovative Diagnosis and Treatment of Chronic Neutropenias,EuNet-INNOCHRON)合作,制定了慢性中性粒细胞减少症的分类、诊断、监测和管理指南,近日发表于《Hemasphere》。基于证据和共识,指南描述了慢性中性粒细胞减少症患者(包括特殊情景,如妊娠和新生儿期)的定义和分类、诊断和随访的指南;特别强调了将临床结果与典型和新型实验室检查以及晚期生殖系和/或体细胞突变分析相结合,对于表征、风险分层和监测整个中性粒细胞减少症患者的重要性,希望这些建议在临床上被广泛使用,使患者、家庭和医生受益。
现将全文翻译如下供各位老师参考,水平有限敬请谅解;如需全文可联系赵龙飞,微信15203118065。

该指南有5个主要主题:(1) 中性粒细胞减少症的定义和分类,(2) 诊断方法,(3) 自然史和随访,(4) 治疗, (5) 特殊情况,并为每个主题制定了一些关键问题。
中性粒细胞减少症的定义和分类
中性粒细胞减少症的定义
中性粒细胞减少症的定义因患者的种族和年龄而异(表1)。在白人新生儿和≤1岁婴儿中,中性粒细胞减少定义中广泛接受的中性粒细胞计数(ANC)临界值为1.0 × 109/L;但是分娩后72-240小时内的足月/近足月新生儿的中性粒细胞减少定义为 ANC 临界值2.5 × 109/L,早产新生儿的 ANC为1.0 × 109/L。新生儿中性粒细胞减少症的更详细特征见后文的“具体情况”章节。从1岁至成年期,中性粒细胞减少症的临界值为1.5 × 109/L。对于白人成人,根据世界卫生组织和 MDS/AML 专家组,采用 ANC 临界值1.8 × 109/L定义中性粒细胞减少症。

需要注意的是部分非洲和中东后裔个体ANC正常范围为0.5~1.5 × 109/L,偶见更低。这种变异以前被称为种族性中性粒细胞减少症,通常作为常染色体隐性性状遗传,与非典型趋化因子受体-1(ACKR1) 基因启动子区 GATA BOX的多态性 (rs2814778,-46T>C) 相关,也称为趋化因子的 duffy 抗原受体 (DARC)。在纯合性 (C/C) 中,该多态性导致红细胞上特异性不表达 Duffy 抗原,该表型被称为Duffy-null。GL-WG 建议引入术语 ACKR1/DARC 相关中性粒细胞减少症 (ACKR1/DARC-associated neutropenia,ADAN),而非种族中性粒细胞减少症,以强调该疾病的遗传而非种族基础。
中性粒细胞减少症的分类
中性粒细胞减少症导致的细菌感染的风险和结局取决于患者招募和将中性粒细胞递送至组织的能力,而不仅仅取决于外周血 (PB) 中的ANC,但临床上没有经验证的方法来估计全身中性粒细胞计数,因此仍外推接受化疗的患者中循环 ANC 和感染之间的定量相关性:ANC 介于1.0和1.5(成人为1.8)×109/L之间时,中性粒细胞减少症分类为轻度;当 ANC 为0.5-1.0× 109/L时为中度;ANC< 0.5× 109/L时为重度;术语粒细胞缺乏症(agranulocytosis)指严重中性粒细胞减少症(ANC<0.2 × 109/L),通常与重度、危及生命的感染高风险相关。中性粒细胞减少症还表现为急性或慢性,取决于持续时间分别为<3个月或>3个月。
除上述传统分类外,GL-WG还认可扩展的、基于发病机制的分类,将中性粒细胞减少症分类为先天性(表2)与获得性和疑似获得性(Likely acquired neutropenia)(表3)及相应的子类。先天性中性粒细胞减少症 (CN) 包括一组遗传性疾病,其特征为骨髓 (BM) 中中性粒细胞的产生、分化和存活受损,对感染易感,以及 MDS/AML 转化倾向增加。CN 可进一步细分:中性粒细胞减少症是唯一异常,中性粒细胞减少症与血液外表现、免疫缺陷/免疫失调、代谢紊乱和营养缺乏相关,或作为更复杂 BM 衰竭综合征的一部分。该分类还确定了每个 CN 亚型的基因。


获得性中性粒细胞减少症可分类为原发性和特发性,与存在抗中性粒细胞抗体或其他未知机制相关,继发于感染、自身免疫性疾病、药物暴露、营养缺乏、脾功能亢进或血液疾病等。疑似获得性中性粒细胞减少症(Likely acquired neutropenia)多为儿童期特发性或自身免疫性中性粒细胞减少症 (AIN),通常具有良性和无并发症的病程,但中性粒细胞减少症24-36个月后仍未消退(持久性),伴或不伴抗体,在3岁后出现(迟发性)。其中一部分患者由于 B 细胞和NK细胞亚群低下而表现出淋巴细胞减少,免疫缺陷/免疫失调疾病相关基因的突变分析已在个体病例中确定相关致病变异。GL-WG认可该组儿童或青少年患者(暂归为获得性特发性)需要进行全面检查以排除潜在的遗传原因。
诊断
患者/家族史中哪些比较重要?
中性粒细胞减少症患者检查的第一步是详细的既往病史和家族史。患者/家族史内容的建议总结于BOX 1。

一般询问
首次发现中性粒细胞减少的年龄是重要信息;尽管 CN 在成年期很少能诊断,但通常在儿童可早期诊断。既往全血细胞计数 (CBC) 结果可明确中性粒细胞减少的持续时间,并区分急性和慢性中性粒细胞减少。了解中性粒细胞减少症是偶然发现还是急性疾病的一部分也很重要。临床病史应了解感染的频率、类型、严重程度和是否需要住院。询问临床事件如发热、口腔溃疡、咽喉痛/吞咽痛、牙龈炎、鼻窦炎、耳炎、皮肤溃疡和蜂窝织炎、深部组织感染、肺炎发作、胃肠道症状和肛周感染尤为重要。反复感染的周期性模式可能提示周期性中性粒细胞减少症 (CyN)。还应了解既往发热或炎症发作是否与 ANC 自发升高相关,以及如何治疗。
其他病史
如果怀疑CN,病史应包括所有血液外部位受累(典型表现见表2)。如果存在提示基础自身免疫性疾病的症状或可导致继发性中性粒细胞减少症的其他疾病,应详细询问病史(表3)。值得注意的是,许多成人 AIN 患者可能存在多种自身免疫表现(如干燥症状、关节痛和Raynaud病)而不符合自身免疫性疾病的明确诊断标准。此外许多患者还存在慢性疲劳和不适。还应获得慢性病毒感染(如病毒性肝炎或HIV)史。应仔细询问用药情况,包括非处方药、常称为天然补充剂的药物和消遣性药物(recreational Drug);中性粒细胞减少症不仅与患者最近开始使用的药物有关,还与最近停用的药物有关。
家族史
详细的家族史(包括血缘关系和种族来源)极为重要,且应尽可能向后扩展,尤其是家族中其他成员有类似症状或实验室检查结果时。此外了解家庭成员曾是否死于感染或 MDS/AML 也很重要。不明原因的婴儿死亡或流产也可能与 CN 有关。值得注意的是,中性粒细胞减少症的家族史可能是阴性的,因为轻度或甚至中度中性粒细胞减少症在既往血液检查中可能被忽略;并且中性粒细胞减少症在同一家族中可能有不同的严重程度。因此来自家庭成员的 CBC 肯定有助于确诊家族性中性粒细胞减少症。
详细的临床检查应包括哪些?
关于中性粒细胞减少症患者临床检查的建议总结于BOX 2。

基于中性粒细胞减少的严重程度和潜在的发病机制,体格检查可能发现口腔溃疡、牙齿和牙龈异常(慢性牙周炎引起)、急性或慢性鼻窦炎、咽炎或耳炎,或皮肤异常,如色素沉着、部分白化病、皮疹、溃疡或脓肿和指(趾)甲营养不良。疼痛和发热时进行腹部检查可能会关注中性粒细胞减少性结肠炎和腹部脓毒症。还应仔细检查肛周和直肠周区域,以排除梭状芽孢杆菌或其他厌氧菌引起的坏死性蜂窝织炎,因为中性粒细胞减少患者可能由于炎症反应降低而仅有轻微的感染体征或症状。淋巴结肿大、脾肿大和肝肿大可能提示感染或相关基础疾病,如骨髓/淋巴恶性肿瘤和代谢紊乱等。
对于儿童,除检查粘膜、牙龈、牙齿、皮肤、指甲和鼓膜外,还应评估生长和发育情况。ELANE突变常与严重 CN 患者发生牙周炎相关,而HAX1 突变患者中高达30%可能存在神经系统问题。身材矮小、软骨发育不良、骨骼、心脏、泌尿生殖系统异常、浅表静脉可见性增加、肌病、指(趾)甲、毛发或皮肤异常、反复胸部感染引起的支气管扩张体征、肝脾肿大以及畏光、眼球震颤和眼皮肤白化病可能与特定 CN 综合征相关(表2)。
对于成人,非常重要的是评估与可能的基础自身免疫性疾病或其他中性粒细胞减少相关疾病相关的体征,如皮疹、关节炎、白癜风、肝脾肿大、眼或口干燥和淋巴结肿大等。
在初始和进一步检查中应包括哪些检查?
急性中性粒细胞减少症患者,尤其是存在感染症状/体征的患者,可能需要立即检查甚至住院,具体取决于中性粒细胞减少症的严重程度和症状。对于不存在潜在 CN 综合征表现的慢性、孤立性中性粒细胞减少症患者,建议使用图1所示的基本检查流程图。
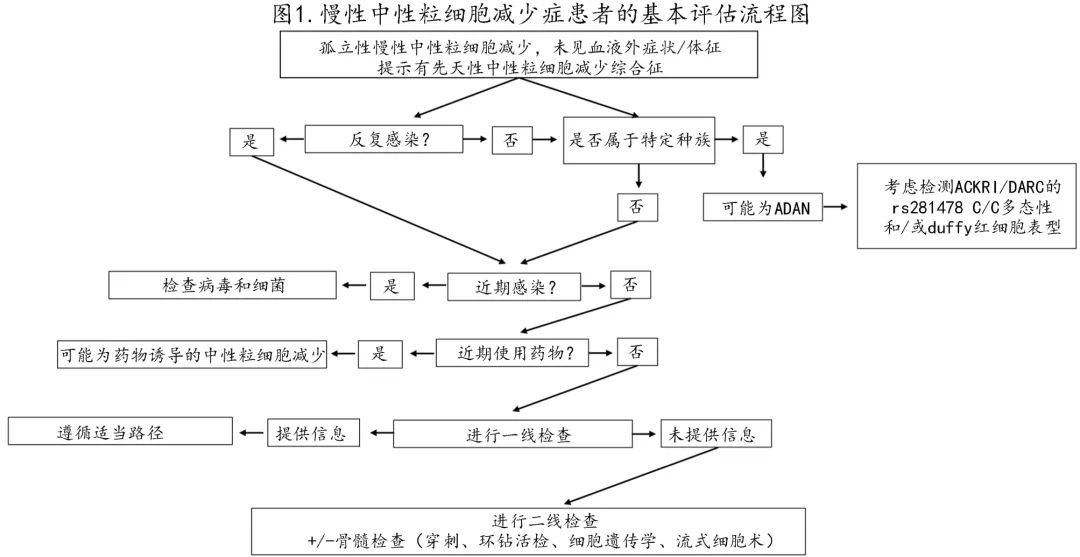
如果初始评估未提示ADAN,也未提示感染后或药物诱导的中性粒细胞减少,则如框3所示进行一线评估(根据所建议检查的可及性,进行调整)。如果一线检查结果未确定,应进行二线评估,如BOX 3所示。

值得注意的是,GL-WG专家强烈建议,如果二线检查结果不确定,儿童、青壮年和特定成人应进行基因检测以排除CN。此外对于有重度中性粒细胞减少家族史,或有典型先天性异常或反复重度感染的幼儿,应在一线检查后进行基因检测。
BM检查的作用是什么?何时和多久一次?
关于 BM 检查类型、时间和频率的建议总结于BOX 4。骨髓穿刺和环钻活检可提供的信息包括:细胞结构、红系、髓系前体、巨核细胞的数量和成熟以及是否存在异型增生特征,还可以告知非骨髓细胞的存在,并有助于鉴别诊断,特别是在成人中。

除形态学外,细胞遗传学评估还可提供关于 MDS 或 AML 的典型获得性染色体异常的更多信息。BM 样本也可用于获得性体细胞基因突变的基因检测,该基因突变在 CN 的白血病生成中起一定作用(如RUNX-1、CSF3R、TP53)或可提示获得性中性粒细胞减少症转化为 MDS/AML 的风险增加(如 SRSF2 和IDH1)。
对于重度慢性中性粒细胞减少症 (SCN) 患者,BM检查可能有助于 MDS/AML 的鉴别诊断和排除。如果在诊断检查期间未进行,应在粒细胞集落刺激因子 (G-CSF) 治疗前给予 BM 检查,以排除MDS/AML,并作为监测恶变风险的基线。在许多 CN 患者中,诊断性 BM 显示中性粒细胞前体在早期阶段(早幼粒细胞/中幼粒细胞)成熟停滞,早幼粒细胞期以外的中性粒细胞系列细胞很少。早幼粒细胞数量略有增加,骨髓嗜酸性粒细胞增多症很常见,细胞结构通常正常或轻微减少,巨核细胞数量和形态正常。如果进行粒细胞-巨噬细胞集落形成单位试验,粒细胞集落的体外生长也可能显示模拟疾病的成熟停滞。
对于原因不明的中性粒细胞减少症成人患者,均应考虑 BM 穿刺和环钻活检结合细胞遗传学和流式细胞术 (FC),以排除 MDS 或其他伴 BM 受累的血液系统疾病(如白血病、淋巴瘤、骨髓瘤和骨髓纤维化),甚至非血液系统恶性肿瘤的BM浸润。对于长期存在、孤立性、轻度中性粒细胞减少症且纵向保持稳定的患者,可以省略 BM 检查,如果进行 BM 检查,如果初始评估未发现任何基础疾病,则无需重复检查,除非 CBC 发生显著变化。
对于 CN 和其他 BM 衰竭综合征,建议每年进行一次 BM 检查(穿刺、环钻活检和细胞遗传学),与 ANC 和 G-CSF 治疗无关。对于未明确为先天性、但需要 G-CSF 治疗的特发性中性粒细胞减少症 (IN),也可考虑BM 检查。
抗中性粒细胞抗体检测的作用?建议进行哪些检查?应该如何解释阳性结果?
抗中性粒细胞抗体在中性粒细胞减少症诊断中的作用和意义的证据主要来自儿科数据。儿童自身抗体的鉴定可验证 AIN 的诊断,AIN是一种典型的早发性良性临床病程、感染少、几年内自发消退、无白血病发生风险的疾病。大多数患者中自身抗体的特异性是针对FcγRIIIb (CD16b)。中性粒细胞减少成人患者中抗中性粒细胞抗体存在的研究远少于儿童,主要靶点似乎仍是FcγRIIIb (CD16b),但与儿童相比抗体特异性更广泛。
检测抗中性粒细胞抗体的最广泛使用和验证的手段是间接粒细胞免疫荧光试验 (indirect granulocyte immunofluorescence Test,GIFT),该方法依赖于将供试粒细胞与患者血清一起孵育,然后使用荧光抗人免疫球蛋白试剂检测结合的抗体。其他检测方法如粒细胞凝集试验和粒细胞抗原的单克隆抗体固定 (MAIGA) 在自身抗体的诊断中作为筛选检测手段劣于 GIFT,但它们可用于测定抗体特异性或作为特定病例的附加方法。基于高通量微珠的检测手段如 LabScreen Multi 已用于同种抗体检测,但尚未在更大系列的 AIN 患者中作为筛查试验进行评估。值得注意的是,大多数临床实验室只使用其中一种方法,根据技术的不同可能有较高或较低的假阳性或假阴性率,因此不应单独使用抗中性粒细胞抗体检测来做出诊断,且应谨慎解读。
鉴定出抗体阳性可使 AIN 的诊断更可信,尤其是在正确年龄组和有典型病史的患儿中。尽管此时不太强调考虑替代诊断,但必须始终考虑假阳性和假阴性结果的可能性;在经基因证实的重度 CN 患者中也有报告抗体检测阳性,因此抗体检测阳性结果不应排除重度 CN。多项研究表明,未检测到抗体但有典型病史的幼儿与 AIN 患者的病程非常相似,在彻底检查排除其他疾病后患者可诊断为IN。IN 和 AIN 具有重叠的临床特征,但前者可能表现为年龄较大、病程较长和不同的免疫学特征,正如本文“分类”部分所述。如果疑似 AIN 但检测结果为阴性,建议在参照实验室重复抗体检测。
在成人,原发性和继发性AIN均可见抗中性粒细胞抗体,但特别重要的是应排除HLAⅠ类同种抗体,它常见于输血患者和既往孕妇,可给出 GIFT 假阳性结果。因此在最终判读阳性抗体结果之前,建议对 HLA I 类抗体进行独立分析,和/或使用 MAIGA 试验或GIFT(基因分型中性粒细胞)确认粒细胞抗体的特异性。
关于自身抗体筛查用于诊断 AIN 的建议总结于BOX 5。

基因检测的作用:哪些基因、方法和细胞来源?它在诊断策略中的位置?
CN 患者的基因检测和分析对于确诊、估计远期并发症(如MDS/AML)相对风险以及为受累患者和家庭成员提供遗传咨询非常重要。由于临床表现的易变性,对于造血干细胞 (HSC) 移植候选者,未受影响的相关供者均应接受基因筛查。
在一线检查结果为阴性后(见BOX 3),所有 SCN 和反复感染和/或有重度中性粒细胞减少症和典型异常家族史的患者,均应通过单基因 Sanger 测序或多基因二代测序 (NGS) 进行基因检查。ELANE突变(约45%的重度 CN 患者中发生)的 Sanger 测序推荐为典型病例基因诊断的首选方法,但家族史或临床结果可能提示需要测序另一个特定的中性粒细胞减少相关基因(表2)。例如在心肌病存在时TAZ(Barth综合征)测序可能具有诊断价值,而存在心脏和泌尿生殖系统畸形时G6PC3测序可能导致诊断。生长不良、吸收不良、脂肪便和骨畸形提示 SBDS 突变见于大多数 Shwachman-Diamond 综合征 (SDS) 患者。总体而言,对疑似基因进行 Sanger 测序相对容易,具有成本效益,鉴定出负责基因(responsible gene)后,还建议使用 Sanger 测序对受累家族成员进行突变筛查。
理想情况下,多基因 NGS 或全外显子测序 (WES) 应包括患者和父母DNA(三人分析)。合理的第一步应包括 CN 中已知突变的所有基因 (>30) 的靶向 NGS panel,可以为所有感兴趣的基因提供统一的测序覆盖,需要较简单的生物信息学分析。检测panel中基因的选择不仅应包括突变时可确切引起中性粒细胞减少症的所有基因,还应包括可导致中性粒细胞减少症为次要特征的疾病的基因(免疫缺陷/免疫失调、代谢和营养缺乏以及其他 BMF 综合征)(表2)。WES 也可用于靶向基因中未发现突变的情况,将分析扩展到基因组中的所有编码区。在所有 NGS 应用中,基因变异致病性的生物信息学解释都具有挑战性,应该基于共识指南进行解读。意义不明的变异体的致病性应尽可能依靠家族分离检查(family segregation study)和功能检查(如相关)。如果高度怀疑CN而NGS panel和 WES 分析均为阴性,应考虑全基因组测序和 RNA测序。应从 PB 中提取用于检测生殖系突变的DNA,但如果 PB 中存在白血病原始细胞,应使用非造血细胞,优选皮肤成纤维细胞、毛囊角质形成细胞或口腔拭子中的细胞(尽管最后一个有血液污染的风险)。随着时间的推移获得的白血病相关体细胞突变可参与白血病转化,可通过指定的 NGS 体细胞组进行检测。除重度中性粒细胞减少症/单核细胞减少症病例外,PB和 BM 是检测髓系特异性基因体细胞突变的等效来源。对于 RNA 测序,应从 PB 或 BM 骨髓细胞中提取RNA,最好是 BM 早幼粒细胞或 PB 中性粒细胞。
对于中东、非洲、印度西部以及也门和埃塞俄比亚犹太人后裔、表现为轻度至中度中性粒细胞减少症(ANC < 0.5 × 109/L的频率较低)且无感染倾向的患者,基因检测也可用于确诊ADAN,准确的基因诊断可节省广泛的非必要检查。该检查可以确定 ACKR1/DARC 基因的 rs2814778 多态性,如本文“定义”部分所述。值得注意的是,在欧洲慢性特发性中性粒细胞减少症 (CIN) 患者(主要是希腊人)中也发现了相同的多态性,因此将 rs2814778 ACKRI/DARC 多态性纳入所有慢性和轻度中性粒细胞减少症患者的检查是合理的。所有上述建议总结见BOX 6。

FC在慢性中性粒细胞减少症诊断中的作用
PB和/或 BM 细胞群的 FC 是诊断特定类型慢性中性粒细胞减少症的支持性工具,如BOX 7所示。

进一步定量不同谱系的细胞群,可以评估细胞表面、细胞内和核内蛋白,以及中性粒细胞和淋巴细胞的生物学功能和免疫特性。FC 在下列中性粒细胞减少相关疾病中特别有价值:
大颗粒淋巴细胞白血病:PB 淋巴细胞 FC 分析对大颗粒淋巴细胞 (LGL)白血病的诊断有重要意义。T-LGL 亚型以CD3+/CD8+/CD57+ 表达为特征,而 NK-LGL 亚型以CD3-/CD8+/CD16+/CD56+ 表型为特征。对于T-LGL,可通过基于 FC 的 T 细胞受体Vβ (TCRVβ) 库和/或基于聚合酶链反应的 TRB 和/或 TRG 基因重排分析确诊克隆性。近期,针对 TRB 基因重排过程中随机选择的2个互斥TCRβ链恒定结构域(TRBC1和TRBC2)中的1个单抗(TRBC1结合单克隆抗体,克隆JOVI−1),被提出作为评估Tαβ细胞克隆性的潜在 FC 标志物。对于NK-LGL,克隆性的评估很难实施;杀伤免疫球蛋白样受体活化亚型的限制性表达可用作单克隆扩增的替代标志物。
骨髓增生异常肿瘤(MDS):FC 可有助于不明原因的中性粒细胞减少患者髓系前体病变或 MDS 病例的识别,具有较高的特异性(高达95%)和敏感性(高达75%)。使用国际 FC 科学学会提出的特定panel对 BM 髓系进行 FC 评估有助于排除MDS。提示 MDS 的髓系异常包括:CD34+ 原始细胞门(blast gate)的细胞的定量和定性改变;未成熟和成熟粒细胞的异常分布;CD11b、CD13、CD33、CD16、CD10、CD5、CD7和 CD56 粒细胞的异常表达模式;基于其侧向散射特征的低粒细胞粒度。FC 还适用于鉴别阵发性睡眠性血红蛋白尿克隆。
原发性免疫缺陷:尽管原发性免疫缺陷的明确诊断多通过遗传分析确定,但 FC 在以下诊断中特别有帮助:(a) 自身免疫性淋巴增生综合征,其特征为TCR-α/β阳性双阴性(CD4-和CD8-)T细胞频率增加;(b) 常见的变异型免疫缺陷,其特征是由于转换记忆 (CD27 + IgD-IgM-)B细胞亚群数量较少,BAFFR突变患者 B 细胞上 B 细胞活化因子受体 (BAFFR) 表达较低,CD19 + B细胞频率较低,ICOS 缺陷患者活化 T 细胞上可诱导共刺激因子 (ICOS) 低表达;(c) 高 IgM 综合征 (HIGM) ,其特征是大多数 X连锁 HIGM 患者中活化的CD4+ T细胞不表达 CD40 配体 (CD40L;CD154),常染色体隐性 HIGM 患者 B 细胞不表达CD40。
端粒生物学疾病:如果可用,建议使用Flow FISH(或其他经临床验证的端粒长度测量技术)排除端粒生物学疾病。鉴于这组疾病无法通过基因检测完全排除,端粒长度测量可能有助于减少诊断缺口。
自然史:随访
哪类患者需要随访(CBC、BM、细胞遗传学和体细胞基因NGS),多久一次?
基线 BM 评估的重要性,包括形态学、细胞遗传学、基于 NGS 的体细胞白血病相关基因突变分析已在上一节中讨论,并总结在BOX 8中。

基线 BM 评估的例外情况如下:(a) 有CN 家族史及已知基因突变的新生儿,可首先进行遗传分析,但不能替代后续的 BM 评估;(b) 抗中性粒细胞抗体阳性且无细菌感染史的婴儿,可以省略 BM 评估,但如果患者发生重度或反复细菌感染或额外CBC异常,应考虑进行 BM 评估;(c)不明原因、长期、轻度和孤立性中性粒细胞减少症且随时间推移保持稳定的成人患者。
BOX 8还总结了后续行动建议。强烈建议每年对 CN 患儿进行 BM 评估,以在发生明显白血病前识别细胞遗传学异常、高危体细胞突变或MDS。应进行 BM 涂片的形态学评估、细胞遗传学分析和基于体细胞 NGS 的白血病基因panel测序,或至少对CSF3R、RUNX1和 TP53 进行深度测序。尽管使用基于 NGS 的 PB 中性粒细胞深度测序已证实 CSF3R 突变检测的高灵敏度,但 BM 形态和细胞遗传学评估对于 MDS 或白血病的诊断至关重要。与 BM 相比,PB中检测其他白血病相关体细胞突变的灵敏度仍在评估中。每年NGS 分析可评估是否获得携带体细胞突变的新 HSC 克隆和相关变异等位基因频率 (VAF) ,还能够监测获得性突变的 HSC 克隆的时间行为。值得注意的是有部分 CSF3R HSC 克隆频率非常高的 CN 患者可数年不发生白血病。此外与同龄的健康个体相比,具有不同遗传背景的 CN 患者可能以更高的频率获得体细胞白血病相关突变(独立于遗传性 CN 引起的突变);获得多种基因病变,尤其是与 CSF3R 突变相关的RUNX1、SETBP1、ASXL1、TP53、PTPN11,可能是中性粒细胞减少期 CN 患者处于白血病前期的强指标,需要更密切的患者随访。
一小部分 CN/AML 患者没有 CSF3R 或 RUNX1 突变但可发生其他致白血病事件。SDS 患者从年轻开始克隆性造血频率便增加,尤其是获得 TP53 和 EIF6 突变时;显性白血病患者可因缺失、CN-LOH或点突变而获得 TP53 位点的双等位基因改变,因此应在 BM 中或在 PB 骨髓细胞(如果 BM 不可用)中进行每年 NGS 白血病易感性骨髓panel分析和 SNP 阵列分析。
对于 CIN 成人,已经强调了 BM 或 PB 细胞基线 NGS 分析对识别髓系恶性肿瘤相关基因突变的重要性(BOX 6)。伴克隆性疾病的 CIN 患者,也称为意义不明的克隆性血细胞减少症 (CCUS) 患者,根据突变的类型和数量以及克隆的大小,转化为 MDS/AML 的风险有所增加。尽管对伴孤立性中性粒细胞减少症的 CCUS 患者的研究有限,但伴任何类型血细胞减少症的 CCUS 患者的报告显示,剪接因子TP53、IDH1/2或DTA(DNMT3A、TET2、ASXL1) 基因突变联合其他髓系基因突变可增加进展为髓系肿瘤的风险。建议携带1个或多个 MDS/白血病相关基因突变的患者,尤其是 VAF 较高 (>10%) 的患者,特别是潜在不确定的克隆性造血频率较低的年轻患者,需要进行更密切的随访(每年> 4次),CBC伴分类计数和 PB 涂片形态学检查。应根据指征(即血细胞减少恶化、大红细胞症和/或形态学异常)进行 BM 检查(包括穿刺、环钻活检、核型和 NGS 分析)。
转化为MDS/白血病的替代标志物
CN 患者恶变为 MDS 或白血病的关键标志如下:PB(假性Pelger-Huët异常、低颗粒、过度分割、网状核和环形核)和BM(有缺陷的肉芽形成、中幼粒细胞阶段成熟停滞和单核细胞形式增加)的典型异常增生特征;细胞遗传学异常;白血病相关驱动基因中体细胞突变的高频率。MDS 期 CN 患者最常见的染色体缺陷为21号染色体三体和7号染色体单体。在 CN 患者中,体细胞 CSF3R 和 RUNX1 突变频率高与白血病发生有明确的相关性。CN 患者典型的白血病相关 CSF3R 突变是受体胞内部分的截断突变,导致1、2或3个酪氨酸磷酸化位点的缺失;RUNX1 错义或截断突变是新发 AML 的特征。在 SDS 患者中,获得TP53位点双等位基因改变与进展为 MDS 和 AML 有关。
在 CIN 患者中,涉及剪接体基因突变的克隆造血和涉及表观遗传调控因子(如TET2、DNMT3A、ASXL1或IDH1/2)的突变模式对 MDS/白血病转化具有阳性预测价值。MDS/AML转化的替代标志物总结于BOX 9。

特殊情况
妊娠
长期 G-CSF 治疗对重度 CN 安全有效,可延长患者预期寿命,因此对于成年后的患者来说,为人父母的愿望已经成为一个新出现的问题。在这方面经常提出两个问题。
是否可以在妊娠期间继续使用G-CSF治疗中性粒细胞减少症?妊娠期间使用 G-CSF 的数据有限,但在所有出版物中,已证实在整个妊娠期间使用 G-CSF 安全且耐受性良好,无明显副作用。
生下受影响孩子的风险有哪些?:随着越来越多的成人患者患有遗传性中性粒细胞减少亚型,受孕前的遗传咨询和母亲在妊娠期间的支持性护理至关重要。重要的是应告知患者,考虑到 G-CSF 治疗的生活质量较高,接受受影响的儿童是合乎情理的。
妊娠期间使用 G-CSF 的建议总结见BOX 10。现有数据支持不同类型 SCN 女性在整个妊娠期间继续 G-CSF 治疗,以预防重大感染和新生儿并发症。该建议是基于重度 CN 患者细菌感染和败血症死亡的高风险;CyN 或 IN 患者的风险可能较低,但由于没有比ANC更好的预测因子,因此建议给予所有ANC 低于0.5 × 109/L的患者 G-CSF 治疗(如果妊娠前接受 G-CSF 治疗);而对于妊娠前未接受治疗的患者,也可考虑 G-CSF 治疗。根据专家的经验,建议在妊娠期间频繁评估ANC,尤其是在 AIN 患者中,因为在妊娠期间可观察到 ANC 升高。

新生儿中性粒细胞减少症
如“定义”和“诊断”章节所述,≤1岁白人儿童的 ANC 正常下限为1.0 × 109/L,但该临界值在出生 < 14天的新生儿中是不同的,其变异性很大,而胎龄是影响正常范围的主要变量。对于6-24小时的健康和足月新生儿,中性粒细胞减少定义为ANC < 6.0-7.0 × 109/L;之后缓慢下降,出生后约72小时限度为<3× 109/L。但早产新生儿的情况差异很大:对于非严重早产儿,ANC下限为出生后24小时 < 3.0 × 109/L,在出生后72小时 <1.0 × 109/L(小于足月新生儿临界值的一半);在极度早产新生儿中,正常 ANC 的第5百分位数在出生后立即约为1.0 × 109/L。此外还有许多其他变量和疾病(与妊娠或分娩相关或不相关)可干扰ANC。与妊娠或分娩无关的变量示例如下:(a) 女性的 ANC 计数平均比男性高2.0 × 109/L;(b) 毛细血管血的 ANC 平均比脐带血高1.5-2.0 × 109/L;(c) 海拔高地的 ANC 平均比海平面高;(d) 新生儿严重坏死性小肠结肠炎,尤其是早产,通常与一过性低 ANC 相关。与妊娠或分娩相关的变量示例包括:(a) 母亲吸烟与 ANC 降低相关;(b) 母亲化疗导致5%-33%的新生儿发生中性粒细胞减少;(c) 母亲抗逆转录病毒治疗导致约20%-50%的新生儿发生中性粒细胞减少;(d) 母亲在妊娠期高血压结果是约一半新生儿的中性粒细胞减少症;(e) 产前生长迟缓是中性粒细胞减少症的独立危险因素;(f) 分娩前劳动导致 ANC 较高;16(g) 无论是否合并严重贫血和血小板减少症,新生儿 Rh 溶血病均与约50%新生儿的中性粒细胞减少症有关;(h) 双胎输血综合征是一种罕见疾病,供者双胎(贫血双胎)中始终存在中性粒细胞减少症;(i)窒息儿中67%存在中性粒细胞减少。
新生儿中性粒细胞减少症的最常见类型如下:
感染引起的中性粒细胞减少症:感染引起的中性粒细胞减少可能是新生儿和婴儿中最常见的中性粒细胞减少。中性粒细胞减少的持续时间通常较短,通常<10天,因此如果在证实感染后数天ANC恢复正常,则诊断为感染相关中性粒细胞减少是适当的。但必须强调,偶见感染(例如由 HIV 或人丙型肝炎病毒引起的感染)可引起持久的中性粒细胞减少。
免疫性中性粒细胞减少症:新生儿免疫性中性粒细胞减少症可细分如下:
1.AIN,在<1月龄时不常见,但并非不可能。
2.新生儿同种免疫性中性粒细胞减少症 (NAN),其中母亲和胎儿之间编码人类中性粒细胞抗原 (HNA) 的基因中的一个多态性的遗传错配导致孕妇免疫、同种抗体通过胎盘和婴儿中性粒细胞减少症。间接抗中性粒细胞抗体在母亲和新生儿中呈阳性。可通过母系血清和父系粒细胞之间的阳性交叉配血获得诊断确认(即使不是常规指征)。虽然约20%的孕妇发现胎母不合,0.6%-1%的孕妇同种免疫,但 NAN 仅发生于1:6000的新生儿
5例 NAN 患者中有1例发生严重感染,主要是皮肤和脐带感染,中性粒细胞减少的平均持续时间为1-4个月。关于治疗的研究不多,但已报告10 μg/kg剂量 g-CSF 的有益作用。关于 G-CSF 治疗的持续时间尚无一致意见,但考虑到中性粒细胞减少仅持续数周,并且有一些严重感染并发症的报告,很可能应考虑 G-CSF 给药直至 ANC 恢复。最后,有少数病例 G-CSF 完全无效,其中部分病例静脉给予免疫球蛋白0.8-1 g/kg有效。
3.继发于母体 AIN 的 NAN 是婴儿早期最罕见的免疫性中性粒细胞减少症。中性粒细胞减少症的平均持续时间与典型 NAN 相同。在报告的患者中有2对兄弟:在首次妊娠时,母亲和新生儿均未接受 G-CSF 治疗,2名婴儿均发生重度感染。在第二次妊娠期间,两名母亲均接受了低剂量G-CSF,两名新生儿均未发生中性粒细胞减少,结局极佳,因此建议向母亲提供G-CSF。
总之,新生儿免疫性中性粒细胞减少症有3种不同类型:(1)NAN:此时证明母亲与新生儿的 HNA 错配以及两者均存在间接抗中性粒细胞抗体是根本;(2)NAN继发于母亲AIN:诊断容易,是受AIN影响的母亲;(3)在<1月龄时出现的AIN:这一情况极为罕见,需要排除假阳性间接抗中性粒细胞抗体,尤其是如果中性粒细胞减少症与并发感染相关,也应进行遗传分析以排除CN。
婴儿一过性中性粒细胞减少症:婴儿一过性中性粒细胞减少症通常发生于在出生后3-4周的早产儿,可能与代偿性红细胞生成和粒细胞生成之间的 HSC 竞争有关。
重度 CN:重度 CN 综合征在新生儿可明显有临床表现。对于在无提示特定疾病/综合征表现的新生儿,应评估孤立性中性粒细胞减少症,建议采用图2流程图中描述的诊断路径。

参考文献
Francesca Fioredda,et al. The European Guidelines on Diagnosis and Management of Neutropenia in Adults and Children: A Consensus Between the European Hematology Association and the EuNet-INNOCHRON COST Action.Hemasphere . 2023 Mar 30;7(4):e872. doi: 10.1097/HS9.0000000000000872. eCollection 2023 Apr.

