Cell Res:北京大学汤富酬团队开发用于单个细胞内多组学分析的长读单细胞测序方法
时间:2023-09-15 23:04:32 热度:37.1℃ 作者:网络
单细胞多组学测序技术为更全面地了解细胞类型基因调控提供了第一步,扩展了系统地探索细胞多样性和异质性的能力。
2023年9月12日,北京大学汤富酬团队在Cell Research在线发表题为“scNanoCOOL-seq: a long-read single-cell sequencing method for multi-omics profiling within individual cells”的研究论文,该研究报道了一种用于单个细胞内多组学分析的长读单细胞测序方法,scNanoCOOL-seq。

在之前的研究中,作者和其他人先前开发了单细胞NOMe-seq(scNOMe-seq)和单细胞染色质整体基因组尺度景观测序(scCOOL-seq)。采用不同的策略,scCOOL-seq/ scNOMe-seq比其他现有的单细胞多组学测序方法有几个优势。然而,基于下一代测序(NGS)平台的分析对长度超过300 bp的DNA片段的表观遗传状态的了解有限。不同的表观遗传特征,如DNA甲基化和染色质状态,可以在相对较长的基因组区域内相互作用,但缺乏同时评估同一单个细胞中多层表观基因组的长读测序方法。
为了制备与纳米孔测序平台兼容的长读文库,亚硫酸氢盐处理的DNA在随机引物时只用一个适配器标记,短片段的两端可能杂交,形成自环DNA,以防止在PCR扩增时被用作模板。这样,得到并比对的读取长度为~900 bp。为了评估scNanoCOOL-seq的可行性,作者对189个HFF1细胞和187个K562细胞进行了分析,分别有70.6%和54.5%的细胞通过了质量控制。原则上,由于GpC甲基转移酶M.CviPI特异性地甲基化可达的GpC位点,内源性DNA甲基化水平可以用WCG的甲基化水平(W = A, T)来表示,染色质可达性可以用GCHs的甲基化水平(H = A, T, C)来表示。平均而言,在单个K562细胞和HFF1细胞中分别检测到974,463个(占总数的3.4%)和821,965个(占总数的2.9%)WCG位点。DNA甲基化水平之间的差异可以通过基因体内部和周围的DNA甲基化谱分析或通过降维。此外,通过scCOOLseq和scNanoCOOL-seq获得的DNA甲基化结果基本一致。
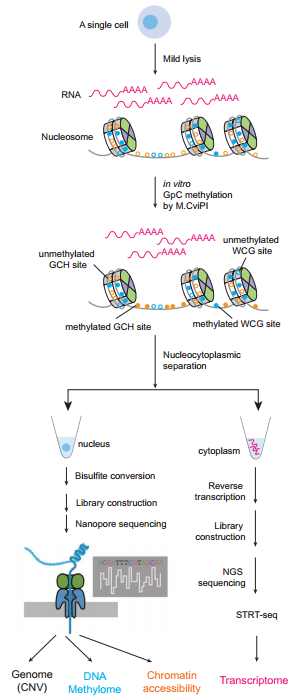
scNanoCOOL-seq方法流程图(图源自Cell Research)
对于染色质可及性检测,作者重点关注转录起始位点(TSS)周围的染色质状态和局部开放的染色质区域,即核小体枯竭区(NDR)。TSS具有高染色质可及性,并且还观察到TSS下游的有序核小体定位模式。使用公开可用的ENCODE数据验证从scNanoCOOL-seq调用的NDR;它们也与基于NGS平台的scCOOLseq获得的结果相当。差异NDR分析可靠地回忆起特异性调节K562或HFF1细胞中基因表达的转录因子。此外,作者观察到启动子可及性与其对应的基因表达呈正相关。这些结果表明,scNanoCOOL-seq在同时分析单个细胞内DNA甲基化和染色质可及性方面表现良好。
综上,该研究开发了一种基于纳米孔测序平台的scNanoCOOL-seq方法,称为scNanoCOOL-seq,它可以联合分析基因组(拷贝数变异(CNVs))、DNA甲基化组、染色质可及性和转录组。利用长读数,scNanoCOOL-seq可以检测全长CpG岛(CGIs)和基因启动子的表观遗传特征,以及具有结构变异(SVs)的基因组区域。例如,可以有效地识别基因组易位位点的表观遗传特征。进一步证明,scNanoCOOL-seq为分析单个细胞内等位基因特异性表观遗传状态提供了很好的机会,例如印迹控制区的等位基因特异性DNA甲基化和染色质可及性状态。总之,该研究开发了一种基于TGS平台的单细胞长读多组学测序新方法。这种方法将在单细胞分辨率上更深入地了解不同分子层的调控关系。
原文链接:
https://www.nature.com/articles/s41422-023-00873-5

