CSRD 2023:Ⅰ型神经纤维瘤病多学科指南解读
时间:2023-09-23 17:13:13 热度:37.1℃ 作者:网络
2023年9月22-23日,由中华医学会、中华医学会罕见病分会主办的中华医学会第一届全国罕见病学术年会(CSRD 2023)于北京国际饭店隆重召开。本次大会邀请国内外罕见病领域知名专家进行专题报告、指南解读、病例讨论、大会发言及论文交流等内容。
本次大会,来自首都医科大学宣武医院吴浩为我们分享《Ⅰ型神经纤维瘤病多学科指南解读》议题。梅斯医学整理重点分享给各位。

国内外已发布多篇指南/共识有助于对Ⅰ型神经纤维瘤的诊疗进行指导
1997年《Ⅰ型神经纤维瘤病护理指南》美国皮肤病学会指南结果委员会
2007年《Ⅰ型神经纤维痛病诊断和个体管理指南》英国神经纤维瘤病协会临床咨询委员会
2020年《基于1966年至今文献综述的神经纤维瘤病Ⅰ法国国家指南》法国PNDS-NF1专家小组
2021年《儿童及青少年神经纤维瘤病诊疗规范》中华人民共和国国家卫生健康委员会
2021年《Ⅰ型神经纤维瘤病临床诊疗专家共识》中国Ⅰ型神经纤维瘤病多中心治疗协作组
2023年《欧洲Ⅰ型神经纤维瘤病患者肿瘤监测指南》欧洲ERN GENTURIS NF1肿瘤管理指南组
2021年我国两个诊疗共识明确指出NF1疾病需要多学科协作诊治,包括《Ⅰ型神经纤维瘤病临床诊疗专家共识》(2021版)和儿童及青少年神经纤维瘤病诊疗规范(2021年版)。
《Ⅰ型神经纤维瘤病临床诊疗专家共识》(2021版):
目前中国尚缺乏对于NF1的诊疗共识,2020年成立中国首个NF1多中心治疗协作组,并进一步联合国内肿瘤外科、肿瘤内科、皮肤科、生殖医学科等学科的知名专家,共同报写了《Ⅰ型神经纤维瘤病临床诊疗专家共识(2021版)》,旨在推进覆盖NF1患者全生命周期的规范化、同质化诊疗,提高中国NF1的诊疗水平和治疗效果。共识强调多学科系统评估、合作诊疗是提高NF1患者治疗水平、生存质量及改善疾病预后等的关键。

儿童及青少年神经纤维瘤病诊疗规范(2021年版):
诊疗规范指出神经纤维瘤病(neurofibromatosis,NF)是一类常染色体显性遗传性疾病,此类族病表型差异性大,以皮肤病变、周围神经系统病变和中枢神经系统肿瘤为主,引起多发的、渐进性的损害,给治疗带来了巨大困难,需要多学科协作诊治。

然而,时隔2年“NF1多学科诊疗指南”再次重磅发布,由中国罕见病联盟Ⅰ型神经纤维瘤病多学科诊疗协作组联合国内专业人士共同制定本指南,旨在提高NF1诊疗水平,为患者提供同质化医疗服务。

本指南涵盖多个科室,覆盖全面。优化NF1患者的诊断和管理需要许多不同科室专科医生之间的密切合作包括但不限于神经内科、儿科、遗传科、眼科、神经外科、骨科、放射科、心内科、皮肤科、内分泌科、肿瘤科、整形外科等。
01 NF1诊断
最新指南NF1诊断标准是依据2021年美国国立卫生研究院(NIH)更新。

Ⅰ型神经纤维瘤病患者随年龄增长可出现多种临床表现或并发症:
色素沉着:咖啡牛奶斑;骨异常:骨发育不良、假关节形成;神经纤维瘤:丛状神经纤维瘤。
3-7岁:学习、认知、社交缺陷:学习障碍、孤僻症、多动症、语言或行动发育迟缓。
7-12岁:骨异常:脊柱异常;神经纤维瘤:皮肤型神经纤维瘤、椎旁神经纤维瘤;胶质瘤:脑干胶质瘤。
18岁及以后:恶性疾病:恶性周围神经鞘瘤、乳腺癌、高级版胶质瘤、嗜铬细胞瘤。NF1患者预期寿命减少约20年。
NF1患者一般出生时即有咖啡牛奶斑(CALMs):
无凸起的大小不一、边界光滑、均匀的色素沉着,颜色自棕褐色至深棕色不等,日晒可使其颜色变深;
99%患儿可出现,是NF1最具特征的临床表现;常在2岁前出现,4岁后新出现的较少,约40%~50%的患者出生时即存在;
80%的患儿年龄每增长1岁,牛奶咖啡斑增加1个,多见于躯干、四肢,以非暴露部位多见;
通常不会恶变,大小及数量与疾病的严重程度无关;范围增大伴过度色素沉着的儿童有更多并发症;
牛奶咖啡斑作为患者首诊时的问题,同时也是大部分患儿早期唯一可能发现的异常。
神经纤维瘤可以分为三种类型,分别为皮肤型、结节型和丛状神经纤维瘤
丛状神经纤维瘤(PN):可存在于30~50%NF1患者中,位于表浅位置并伴有皮肤和软组织过度生长,也可位于体内深部,或者同时分布在浅表和深部。累及多条神经干、分支及神经丛,可侵及周围组织。可引起疼痛、运动功能障碍、毁容和恶变等。出现恶变的最常见特征是病变疼痛且不断扩大”。
皮肤型神经纤维瘤:常呈质软、无蒂或有蒂的肿瘤,无压痛,如加速生长可出现瘙痒症状。通常在青春期前或青春期期间开始出现,大小和数量随年龄而增长。
结节型神经纤维瘤:生长在皮下或体内的离散性病变,一般不会侵袭周围组织。可能有压痛,质硬而富弹性。瘤体增大压迫周围结构可引起疼痛,可转化为MPNST。
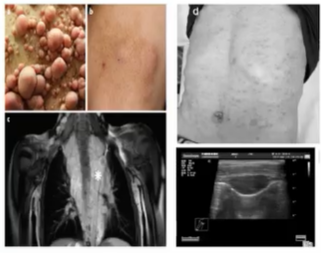
从病理学分类可分为:局限/结节型神经纤维瘤、弥漫型神经纤维瘤和丛状神经纤维瘤
局限/结节型神经纤维瘤:界限分明的真皮/皮下结节,切面发亮,呈棕褐色,累及主要神经时表现为有包膜的梭形肿块。
弥漫型神经纤维瘤:边界不清的真皮/皮下结节,切面呈棕褐色。
丛状神经纤维瘤:伴有多发结节的肿块,宛如“一袋蠕虫”样外观。
结节型和丛状神经纤维瘤也可根据肿瘤部位进行分类
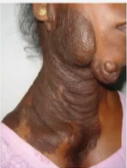
头面部神经纤维瘤:可导致毁容,以及由于感觉器官被肿瘤侵犯而导致相关残疾,例如肿瘤侵入眼眶可导致患者眼球的移位以及视力的丧失。
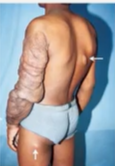
椎旁神经纤维瘤:可导致脊柱受压,从而引起麻痹或瘫痪。
纵隔神经纤维瘤:通过压迫气管或大血管,导致危及生命的心肺功能损害。

位于四肢的神经纤维瘤:由于侵犯周围组织以及静脉瘀滞而导致严重的功能障碍。
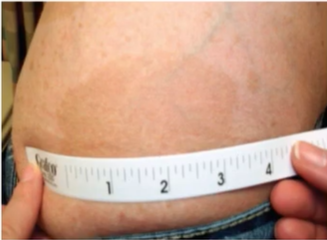
此外,腋窝和腹股雀斑是NF1另一常见的临床特征,颜色与牛奶咖啡斑相同,比咖啡牛奶斑小;通常在3 ~ 5岁时出现,发生率约为90%,常首发于腹股沟区;还可出现于其他易摩擦部位,可表现为弥漫性,是NF1 诊断标准中最特异的表现。
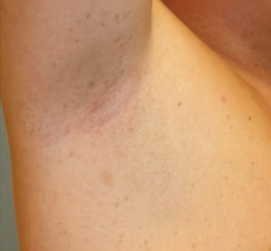
15%的6岁以下NF1儿童可出现视路胶质瘤(OPG)
通常为低级别毛细胞型星形细胞瘤,可出现在沿前视觉通路至视放射的任意位置,累及视神经、视交叉和视交叉后视束;15%的6岁以下NF1儿童可出现OPG,年龄更大的儿童和成人中极少出现;大多数惠儿视力正常,如病变扩大,可引起进行性视力丧失另可表现为色觉下降、瞳孔功能异常、眼球突出和视神经萎缩。

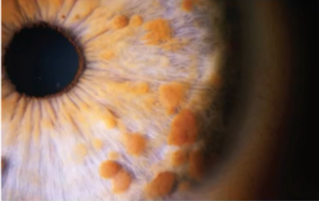
虹膜错构瘤(Lisch结节)是NF1的一种特异性表现
虹膜凸起的棕褐色错构瘤,常见于双侧,呈半球形白色或黄棕色降起斑点,境界清楚的胶样结节,是NF1的一种特异性表现,对视力无影响。通常6岁时30~50%出现虹膜错构瘤(Lisch结节),92%在成年时出现。
结节的数量可能和疾病进展速度相关,因此评估患儿疾病进展最佳应该5岁之后定期通过裂隙灯显微镜检查明确虹膜错构瘤(Lisch结节)数量,而非通过牛奶咖啡斑的改变。
NF1患者的特征性骨病变,包括蝶骨翼发育不良,胫骨前外侧弯曲(胫骨发育不良)或长骨假关节
主要包括骨发育不良和假关节;其他还包括身材矮小、脊柱侧凸、脊椎发育缺陷、蝶骨翼发育不良、非骨化性纤维瘤和骨质疏松等。长骨发育不良在婴儿或年幼儿童中通常表现为胫骨前外侧凸,可进展为髓腔狭窄、骨皮质增厚和骨折,约半数的骨折发生在患者2岁前。在约5%的NF1患者中,长骨假关节出现于婴儿期,男性多于女性(1.7:1)。约10-64%的NF1患者有脊柱侧弯,常发生在6~10岁或青春期早期。

对于临床症状不明显的儿童患者,可以通过NF1基因检测辅助诊断
价值:对于儿童患者,目前的临床诊断标准对儿童的敏感性较低,而体征会随年龄增加而逐渐出现;对于小于7岁的儿童和只有咖啡牛奶斑、皮褶雀斑却没有其他临床标准的患者,可进行基因检测来确认NF1的诊断。
类型选择:目前主流的测序方案为候选基因(NF1)的基因芯片检测和全外显子组测序(whole exome sequencing,WES),如果条件允许,优先推荐先证者和/或父母行三人全外显子组测序(Trio-WES);随着全基因组测序(whole genome sequencing,WGS)技术的成熟以及费用的降低,条件允许可行WGS。
结果解读:在基因报告中,检测出NF1基因的致病性(pathogenic,P)和可能致病性(likely pathogenic,LP)变异,可明确NF1的诊断;对意义未明的变异(variants of uncertain significance,VUS),可结合患者的基因型-表型关联、RNA测序等多组学数据进行遗传分析;NF1患者基因检测结果为阴性时,可重点关注剪切变异、非编码区变异和同义变异进行定期重分析。
推荐意见:应对临床诊断不明确,又需进一步诊疗或遗传咨询的疑似NF1的患者进行合适类型的基因检测以明确分子诊断、辅助制定疾病管理方案;首选方案为全外显子组测序,根据MF1的不同类型选择不同的送检样本;若检测结果仍为阴性,可考虑采用全基因组测序,并且每两年对原始数据进行重分析,以纳入新发现的NF1相关的基因或新突变等。
基因检测有助于明确鉴别NF1与其他类似综合征
NF1患者诊断时需与其他类似综合征鉴别,包括Legius综合征、McCune-Albright综合征、2型神经纤维瘤病、Noonan综合征和结构性错配修复缺陷综合征等。

诊断的过程中建议NF1患者应用合理的影像学手段辅助筛查、诊断
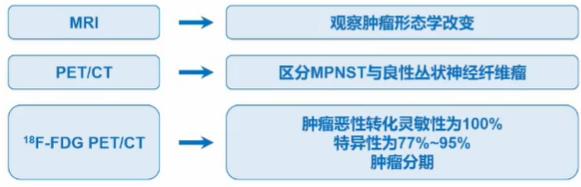
应根据病史和体格检查结果决定是否进行影像学检查。当出现新发症状或体征时,若丛状神经纤维瘤患者出现症状,例如进展性剧烈疼痛、肿瘤生长迅速、结节样突出,需行影像学检查,推荐的影像学检查方法为增强MRI、PET/CT。
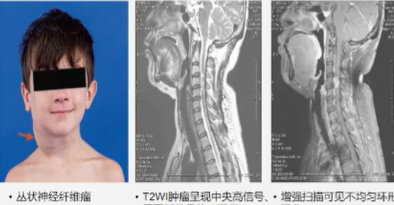
MRI可诊断结节型和丛状神经纤维瘤
神经纤维瘤在MRI T1W1上无明显特征,T2WI上是高信号,信号多不均匀,可呈中央低信号,周围高信号,称之为“靶征”,增强扫描明显强化,多不均匀。
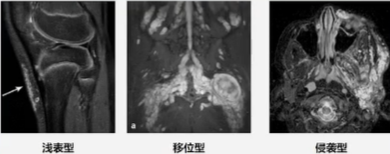
Friedrich于2003年首次提出基于磁共振成像(MRI)的神经纤维分类方式:
- 浅表型:仅限于皮肤和皮下组织、不侵犯筋膜和肌肉的非侵袭性肿块;
- l移位型:位于皮肤深层或体内的多结节肿块,可以因体积巨大压迫邻近结构,但不会侵入相邻的肌肉或皮肤;
- l侵袭型:不可分割的团块状肿瘤,存在肌肉、筋膜、关节和周围组织的侵犯,与周周正常组织边界不清。
02 多学科治疗方案
NF1患者可根据不同的皮肤症状选择不同的治疗方式
咖啡牛奶班(CALMs):目前尚无针对NF1患者的CALMs系统治疗研究;主要采用对症处理,如皮损严重妨碍美容,可采用激光治疗(如调Q激光,其治疗原理是选择性光热作用,调Q激光通常不引起瘀痕,但疗效反应个体差异较大)。
推荐意见:NF1患者的CALMs一般不必处理,对严更妨碍美容的皮损,可尝试激光治疗。

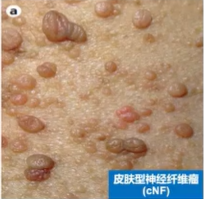
皮肤型神经纤维瘤(cNF):手术切除是cNF治疗的主要手段,术后并发症包括局部感染、瘢痕、复发等。
推荐意见:皮肤型神经纤维瘤为良性肿瘤,治疗需结合患者意愿,对较大或对躯体功能造成影响的瘤体采取手术为主的治疗;同时,可采取激光消融、电干燥术、激光光凝术及射频消融术,用于治疗瘤体数量较多严重影响外观的cNF患者
结节型和丛状神经纤维瘤的手术治疗难度大,需多学科团队共同制定手术方案
1、手术目标:对于体积巨大和/或浸润性肿瘤,考虑到其良性性质,手术策略应是切除肿瘤以缓解症状,同时保留神经功能,对该类型肿瘤进行成形手术,通过缩小肿癌以恢复社会接受度更高的外观。
2、手术指征:推荐对产生临床症状的神经纤维瘤、具有恶变影像学证据的病灶,以及体积过大的肿块(直径> 6cm)进行手术。
3、手术方案:应根据手术需要与包括银科医生、神经外科医生在内的多学科团队一起制定手术方案。
4、手术挑战:部分神经纤维瘤,尤其是巨大、侵袭型瘤体,具有广泛累及正常组织、术中易出血、累及重要神经等特点,增加了术中出血、术后神经功能障碍等并发症的发生率,给手术操作带来巨大难度。
丛状神经纤维瘤患者手术术后有复发风险,需要长期规律随访
推荐意见:对于丛状神经纤维瘤的患者应进行长期、规律随访,并依据年龄、肿瘤部位、手术范围等对患者进行分级管理、肿瘤监测,指导手术的开展及复发风险的评估。
丛状神经纤维瘤术后复发较为常见,复发概率在25%~66%不等;
复发或再次进展与多种因素相关,包括切除范围、患者年龄、肿瘤位置和生长类型;
手术切除范围越小、患者手术年龄越小,术后复发进展的风险越高;
不同部位的丛状神经纤维瘤表现出不同的术后复发率,头部/颈部/面部肿瘤进展率最高,躯干部次之,四肢部位复发率最高;
推荐术后对丛状神经纤维瘤患者进行长期、规律随访,应依据年龄、肿瘤部位及手术范围将患者进行危险分层,有助于指导手术开展及复发风险评估。

MEK抑制剂可作为丛状神经纤维瘤患者的药物治疗选择
推荐意见:根据患者瘤体情况及个人意愿选择手术治疗及MEK抑制剂靶向治疗。
靶向治疗:司美替尼:一种可诱导肿瘤缩小的口服选择性丝裂原活化蛋白激酶(MEK)抑制剂;2023年4月28日,科赛优(硫酸氢司美替尼胶囊)获批,在中国上市;适用于3岁及3岁以上伴有症状、无法手术的丛状神经纤维瘤(PN)的Ⅰ型神经纤维瘤病(NF1)儿童患者。
其他治疗方案:除MEK抑制剂之外,另有几类靶向疗法在临床试验中表现为对丛状神经纤维瘤有效,例如多酪氨酸激酶抑制剂;一些潜在的治疗方法,包括基因治疗、免疫疗法等,目前仍处于研究阶段。
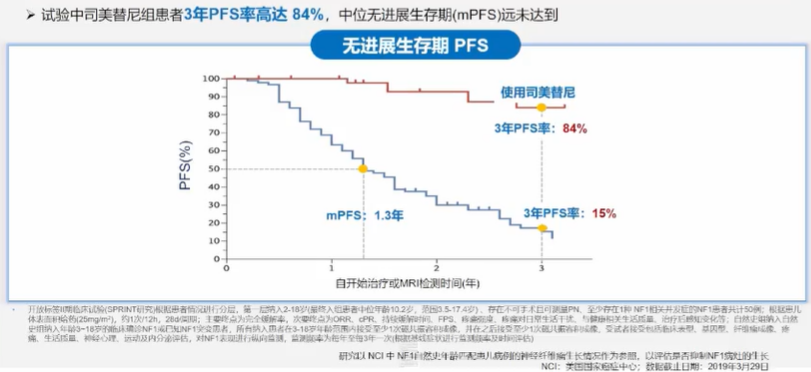
司美替尼治疗3年PFS率高达84%,相比自然史患者获益明确
各药物目前在研现状

03 NF1患者如何进行全周期健康监测
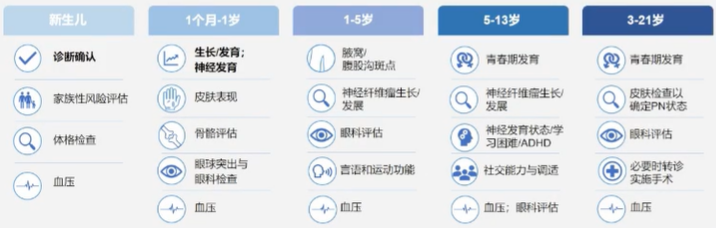
为优化神经纤维瘤的疾病管理,NF1患者需根据年龄进行个体化检查
NF1患者的体格检查项目各年龄段均有不同,一旦发现残留病变再生长或有新病变发生,应高度重视,若引起新的明显畸形或功能障碍应考虑再次干预。
因NF1患者起病年龄不一,临床表现多样,指南新增推荐NF1患者需接受全周期健康监测
由于NF1患者起病年龄不一,临床表现多样且受累系统较多因此NF1患者在各个阶段都应接受定期健康观察监测,以评估疾病进展,及早干预。
诊断初期,根据患者病情诊断需要可选择合适类型的基因检测,必要时随访期间间隔2~3年进行数据重分析。
若有生育意愿,应至少在生育前进行一次产前遗传学咨询。
青春期前儿童,应每年进行1次头围测量评估,从5岁起每年进行1次性早熟监测至青春期性发育开始。
儿童或青少年起病患者,均应在儿童期和青春期期间接受1次发育和心理评估。
对于所有年龄起病的NF1患者,建议全周期健康监测应包括:眼科检查、骨科检查、皮肤检查、心血管系统、乳腺筛查、神经系统。
重视关注患儿心理健康,进行恰当的疏导和管理也至关重要
NF1患儿最常见的精神障碍:焦虑障碍、语言障碍、孤独症谱系障碍、抑郁障碍、注意缺陷多动障碍、社交障碍、睡眠障碍、学习技能障碍、遗尿症等。
进行针对性的心理护理,实施有效的心理疏导、技巧性沟通,帮助NF1患者调整心理状态:
- 生活上主动关心体贴患者,增强社会支持,发挥家庭成员的积极作用,让患者得到情感支持;
- 治疗上让患者满意放心,提高患者的心理承受力,以良好的心境接受治疗;
- 治疗后加强心理护理,增强患者生活的信心,提高生存质量。
对于NF1患者来说,即使没有已知或可见的PN,也要重视PN筛查及长期的随访监测
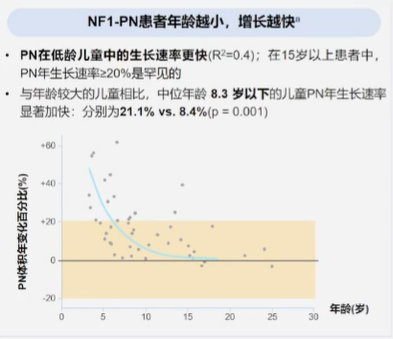
自诊断或出生开始,应对患者PN风险进行监测:
- 可通过观测,触诊和神经系统检查对患者进行临床评估,影像或视频是有效的辅助手段(证据等级:中等)
- 应从诊断或出生时即开始PN的临床评估,并在每次就诊时重复进行(证据等级:中等)
- 从儿学期到成年期,至少应进行1次全身MRI成像(WB-MRI)以监测PN,并评估患者MPNST的进展风险。对MPNST高危患者,可进行更高频率WB-MRI评估(证据等级:弱)
- 重复成像的频率应基于患者的个体情况进行确定,对MPNST高危患者可考虑增加频率。若未发现MPNST,则仅需进行临床评估(证据等级:中等)
- PN的临床监测应在首次发现并在每次就诊时重复进行(证据等级:中等)
- 有症状的PN患者需要增加对ANNUBP/MPNST的监测。活检之前,在诊断过程中可使用18FDG PET MRI(首选)或18FDG PET CT(若18FDG PET MRI不可及)结合临床评估和MRI(证据等级:中等)
- 对于有症状的PN,手术是目前准一有可能治愈的方法,应考虑进行PN手术(证据等级:中等)
- MEK抑制剂可考虑作为有症状的PN和不可手术的有症状的PN的治疗选择(若是国家标准护理的一部分)(证据等级:中等)
- PN的治疗应由具有NF1专业知识的多学科团队来决定和进行(证据等级:弱)
- 考虑NF1相关PN患者有潜力的恶化风险,在进行管理决策时应提供心理支持(证据等级:弱)
丛状神经纤维瘤有恶变风险,需长期随访监测
NF1患者发生恶性周围神经鞘瘤(MPNST)的终生风险在8%到13%之间,通常出现在已存在的丛状或结节性神经纤维瘤内,MPNST好发于20-50岁成人。
患病风险:
- 非体表的PN且数目较多、瘤体较大、年龄较轻、整个NF1基因缺失的NF1患者;
- 当原有的肿块大小或疼痛出现明显变化,或神经功能障碍迅速进展时,应警惕发生恶性转化,尤其是肿瘤大小的变化最能预测肿瘤的恶性程度。
- MPNST好发于颈部或四肢的大神经干上,尤其是周围神经干,如臂丛神经、骶丛神经、坐骨神经等。
MRI出现瘤体体积较大、瘤体累及筋膜层深面及坏死,提示恶变可能;
MRI对于MPNST的诊断、临床分期、治疗及预后评估方面具有很高价值, 为其首选的影像学检查手段;
对于肿瘤负荷重且具有MPNST高风险的患者,可定期接受MRI和/或PET/CT筛查。
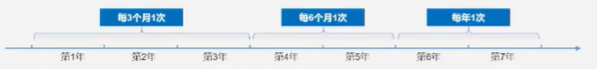
为更好的实现NF1患者的全周期健康监测建议磁共振成像(MRI)应用于NF1患者全病程
诊断:患者一旦出现并发症,能及早发现并对症治疗;当出现新发症状或体征时,推荐的影像学检查方法为增强MRI、PET/CT,MRI可观察肿瘤形态学改变。
治疗:结节型和丛状神经纤维瘤手术术前准备,建议术前通过MRI、CT及PET等影像学检查充分评估瘤体的生长模式、累及范围、良恶性及全身脏器受累情况。
监测:丛状神经纤维瘤常规监测手段是MRI,MRI扫描可显示丛状神经纤维瘤的大小和严重程度,可作为定期评估丛状神经纤维瘤生长状态的常规监测手段,NF1患者应每年常规行脊柱MRI检查。
指南新增多学科诊治流程,为患者及家庭提供长期的、个体化、综合性疾病诊疗随访咨询服务
多学科诊治流程总结:NF1作为一种罕见疾病,且受累系统广、表现多样,NF1患者在普通门诊常被误诊;NF1患者在进入成年期后也常面临随访和管理的困境;建议在有条件的中心设立专门的NF1多学科诊治平台,为患者及家庭提供长期的、个体化、综合性疾病诊疗随访咨询服务。

NF1疾病知晓率极低,且患者临床表现较为多样化。面对NF1,不是神经科一个学科能够应对的,需要多学科参与,包括皮肤科、外科、骨科、眼科、遗传科、药剂、护理、心理等,共同围绕着疾病所累及的脏器开始整体性地评估、诊断、治疗、管理、干预、康复。
另外,NF1作为一种有药可治的罕见病,MDT模式(多学科联合诊疗)非常重要,凝聚和培养MDT团队,提升疾病诊疗水平和研究水平,结合创新药司美替尼,攻克Ⅰ型神经纤维瘤,相信指日可待。
总结
NF1患者具有临床表现复杂,需要多学科协作对患者进行疾病评估,并制定多学科诊治方案。药物治疗为NF1患者提供了新选择,司美替尼被推荐用于3岁及3岁以上伴有症状、无法手术的丛状神经纤维瘤(PN)的Ⅰ型神经纤维瘤病(NF1) 儿童患者的治疗。NF1患者起病年龄不一,临床表现多样且受累系统较多,NF1患者需要长期规律随访,在各个阶段都应接受定期健康观察监测,以评估疾病进展,及早干预。

