Nature子刊重磅综述:急性淋巴细胞白血病(流行病学和机制部分)
时间:2024-06-18 18:01:40 热度:37.1℃ 作者:网络
急性淋巴细胞白血病 (ALL) 是一种以未成熟淋巴细胞不受控制的增殖为特征的血液系统恶性肿瘤。在过去的几十年里,人们在理解 ALL 的生物学方面取得了重大进展,使其诊断、治疗和监测得到显著改善。自化疗出现以来,ALL一直是一个合适平台,可用于测试适用于一般癌症的创新方案。例如,组学医学的出现使人们对支持 ALL 的分子和遗传特征有了更深入的了解,基因组分析技术的创新也确定了驱动 ALL 的特定基因改变和突变,激励了新疗法。靶向药物(如酪氨酸激酶抑制剂和免疫治疗)在患者亚组中显示了有前景的结果,同时将不良反应降至最低。此外,CAR- T 细胞治疗的发展代表着 ALL 治疗的突破,可带来显著缓解和潜在的长期缓解。其进展并不仅限于治疗方式。可测量残留疾病(MRD)监测和离体药物反应谱筛查可实现疾病复发的早期检测和卓越应答者的识别,使临床医生能够调整个体患者的治疗策略。除此之外,数十年的支持和预防性治疗也改善了治疗相关并发症的管理,提高了 ALL 患者的生活质量。
《Nature Reviews Disease Primers》近日发表长篇综述,讨论了 ALL 的流行病学、机制和诊断,以及 ALL 的最新进展,强调了在改善患者结局和生活质量方面的创新方法。文章较长,现分两部分翻译,水平有限如有错误敬请谅解。

引言
急性淋巴细胞白血病 (ALL) 是最常见的儿童恶性肿瘤,是儿童和青少年(<19岁)疾病死亡的主要原因,也是成人癌症相关死亡的重要原因,2019年全球有>15万例新发病例。ALL 源于 B 细胞前体或 T 淋巴祖细胞的转化和不受控制的增殖(BCP-ALL、T-ALL)。B淋巴或T淋巴祖细胞(淋巴母细胞)在淋巴细胞分化的几个未成熟阶段中的任何一个阶段停止,增殖并取代骨髓和外周血的正常造血细胞。ALL 表现为骨髓和其他外周器官(如肝脏、脾脏和淋巴结)的原始细胞浸润,导致白细胞 (WBC) 计数升高、红细胞和血小板计数降低(全血细胞减少症)以及中枢神经系统(CNS)受累(CNS也可能是疾病复发的部位)。诊断时,BCP-ALL很少表现为淋巴母细胞淋巴瘤 (LBL);Burkitt淋巴瘤偶尔表现为 IV 期骨髓受累(原始细胞>25%),与白血病一致。T-ALL 常表现为淋巴瘤特征,如淋巴结或结外发病,包括胸腺肿大。
基因组研究揭示出 ALL 的高度异质性,对其分类提出了挑战。基于此,第5版 WHO 血液淋巴组织肿瘤分类 (WHO-HAEM5 2022) 支持两个组织学亚组的差别,同时引入以分子事件定义为中心的新命名法,如BCR::ABL1融合,而非相应的细胞遗传学改变,如t(9;22)(q34.1;q11.2)。例如,基于基因表达和测序技术的进展,WHO-HAEM5 引入两个新的亚型:BCP-ALL/LBL伴ETV6::RUNX1样特征和BCP-ALL/LBL伴TCF3::HLF融合。同时,它也承认 BCP-ALL伴BCR::ABL1样特征和 iAMP21-ALL 两个临时亚型为不同类别。T-ALL 的子分类没有显著变化,包括T-ALL非特指型和早期 T 细胞前体急性淋巴细胞白血病/淋巴瘤 (ETP-ALL)。同年,国际共识分类 (ICC)制定了略有不同的 ALL 分类,包括 WHO-HAEM5 中未涵盖的临时亚型和新亚型,强调了驱动结构病变或独特的基因表达特征。
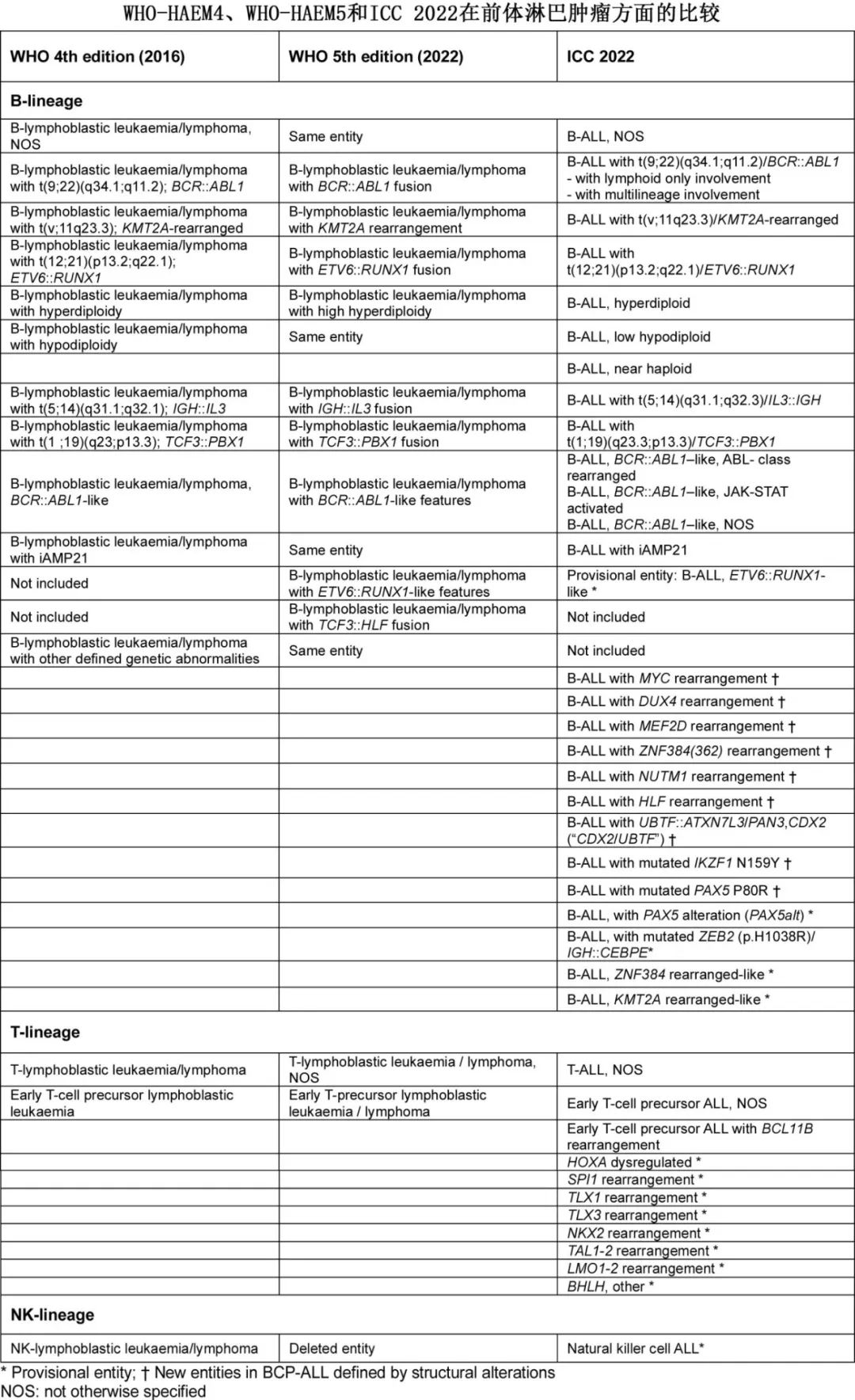
纵观现代历史,ALL都是测试先进技术和新疗法的平台。研究人员首次在这类肿瘤中精确地用化疗药物诱导疾病缓解(下图)。人工合成的叶酸拮抗剂4-氨基喋呤酰谷氨酸 (aminopterin) 是首个可诱导ALL 患儿缓解的药物,它是现代化疗的标志性经历。随后的改进包括越来越复杂的风险调整综合化疗伴加强抗菌预防、省略预防性头颅照射和监测可测量残留病灶(MRD;既往称为微小残留病),它们均取得超越想象的成功,尤其是在儿童(0-14岁)和青少年(15-19岁)中,5年相对生存率分别为92%和77%,毕竟早期单一化疗药物仅可获得暂时缓解或疗效有限。
所有研究都包含许多技术进步。例如,详细的细胞遗传学特征和基因表达谱能够根据复发基因重排或癌基因异常表达改变来区分肿瘤亚型。二代测序 (NGS) 有助于在疾病的各个阶段识别复发突变及其克隆进化,从而促进靶向治疗的开发,增进对耐药机制的理解。此外, CAR-T 细胞治疗和基于抗体的免疫治疗的有效性已在 ALL 中得到验证,为传统化疗无效或复发的患者提供了额外的选择。
流行病学
挑战
由于各种因素,包括获得医疗保健系统和最新诊断技术,尤其是在低收入国家,确定 ALL 的全球发病率具有挑战性。最可靠的癌症登记,如国际癌症研究机构 (IARC)、全球癌症观测站 (GLOBOCAN) 和国家癌症研究所 (NCI) 监测、流行病学和最终结果 (SEER),以及各种国家癌症登记,倾向于优先考虑特定地理区域。大多数流行病学研究主要关注高收入国家,如美国和欧洲。
发病率、患病率和年龄分层
ALL占所有新诊断癌症的3%左右,占癌症相关死亡率的3%,且在年龄、性别和地区方面分布不均(图1)。随着WHO分类的不断演变,自1990年以来,ALL的发病率增加了1.29%,这要归功于改进的诊断,从而发现了新的亚型和变体。由于T-ALL发病率的男女比例为2:1,男女比例介于1.22和1.41之间,这可能是由于性别特异性肿瘤抑制基因;男性的年龄标化发病率略高于女性。
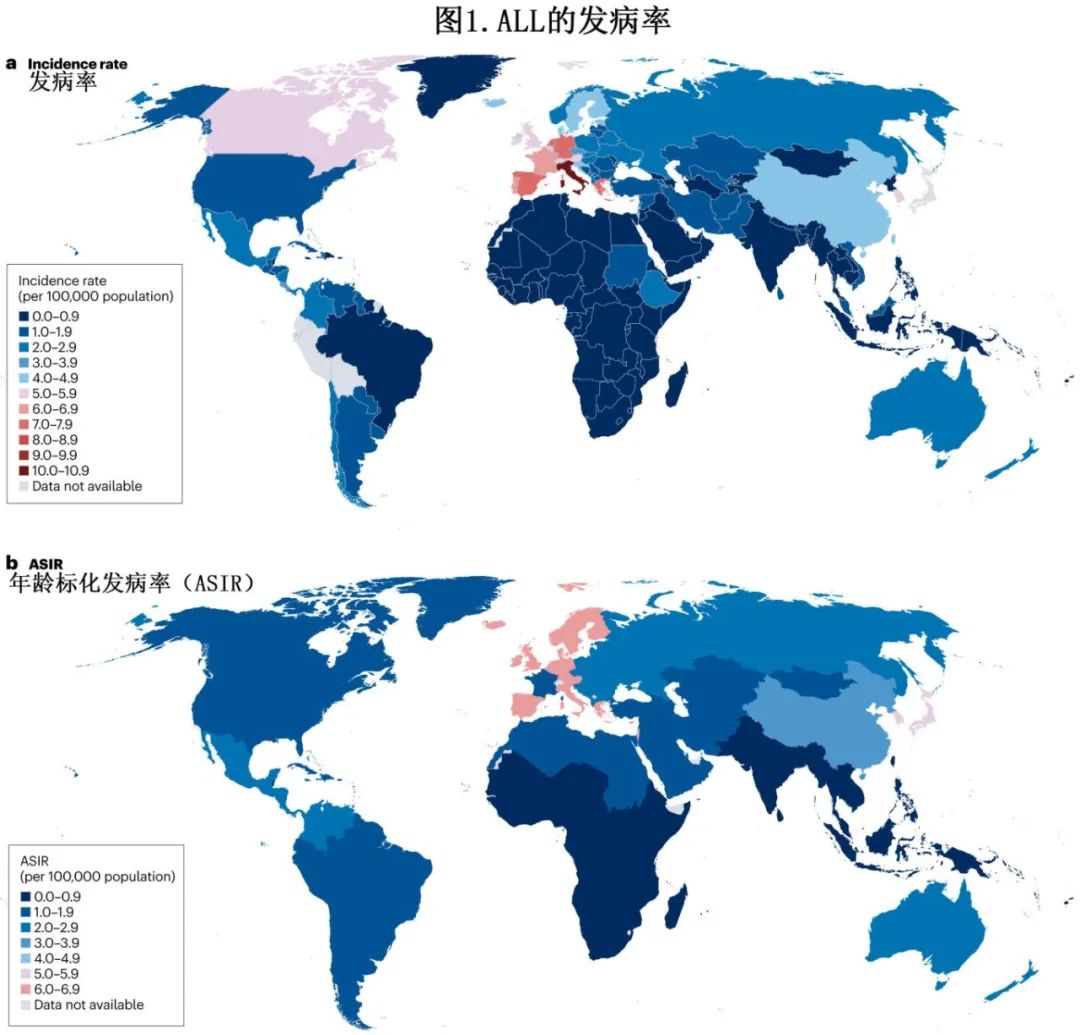
如果考虑社会人口指数(SDI),ALL的发病率在中-高SDI的国家中较高,这些国家的年龄标化发病率增长最快。在全球范围内,ALL发病率最高的是出生至9岁的患者。ALL在老年人群(69岁以上)中的发病率也在增加,可能与全球人口年龄的增长有关。在美国,西班牙裔人群的ALL发病率最高。
在ALL亚型中,BCP-ALL占80%,约23%的成人ALL为费城染色体(Ph) (BCR::ABL1)阳性;在B系白血病谱系中,Burkitt白血病占1-2%。T-ALL占所有病例的20%。
死亡率和残疾
尽管2019年的年龄标化死亡率较低,为每10万人0.63,但ALL的全球死亡率已从1990年的41240人增加到2019年的47650人。中拉丁美洲(每10万人1.72人)和安第斯拉丁美洲(每10万人1.26人)的死亡率较高,其中男性患者的死亡率较高。在SDI高-中等或中等地区,ALL死亡率更高(年龄标化死亡率0.70-0.74)。2019年,中国和印度的死亡人数最多,分别超过1.1万人和4000人。在中等SDI国家,特别是拉丁美洲和东亚,ALL死亡率变化的估计年度负担百分比有所增加,而高SDI国家,特别是澳大拉西亚,则大幅下降。2020年,美国报告的ALL死亡率为每10万人0.4人,高龄组(65岁以上)死亡率更高。由于过早死亡而损失的生命年数和由于残疾而损失的健康生命年数(残疾调整生命年,DALY)的危险因素包括男性、中等SDI地区、高SDI地区女性的甲醛暴露和吸烟。尽管从1990年到2019年的30年间,ALL的DALY总体减少了6.9万,但ALL治疗的临床需求仍未得到满足,包括T-ALL等亚型、复发难治性(R/R)和老年患者,因此需要创新疗法来提高治愈率。
危险因素和易感性
已经确定了ALL的多个危险因素和易感条件。白种人、男性、有兄弟姐妹被诊断患有白血病、暴露于电离辐射、化疗或放疗以及暴露于霉菌毒素和苯等碳氢化合物在内的毒素,均为ALL的潜在危险因素。吸烟、电磁场、酒精和药物与ALL的相关性较弱或不一致。
在白血病易感性和发展的分子学基础方面,已对儿童 ALL 进行广泛研究。尽管病因在很大程度上未知,但仍出现许多假设,包括外源性和/或内源性毒素暴露、生殖系易感性和随机事件。也有人认为感染具有作用,并提出两个主要假说。第一个假说是基于,观察到儿童 ALL 的起始事件(即“首次打击”),如高超二倍体ALL、ETV6::RUNX1、BCR::ABL1、TCF3::PBX1和KMT2A 重排,在子宫内产生于一组造血细胞(携带这些突变但未进展至全面白血病阶段)的克隆扩增,也称为白血病前克隆。在少数携带首次感染白血病前克隆的个体中,出现了一种独特的情况:婴儿时期缺乏接触感染的机会,再加上由于应激性感染(称为“二次打击”)而在出生后引发的遗传异常,作用于儿童早期新生的、相对不发达的免疫系统。这种复杂的相互作用称为Greaves‘延迟感染’假说,或可促进儿童ALL(图2a)。第二种假说是基于一种寻求解释 ALL 流行集群的流行病学方法,称为‘人群混合’假说 (图2b)。在这种情况下,ALL被认为是由于人口混合增加而暴露于新病原体的结果。当一大群人,包括许多来自城市背景的人因此暴露于各种感染时,这种影响尤其明显,他们搬迁到人口稀少的地区,而当地人口的很大一部分事先没有接触过这些病原体。
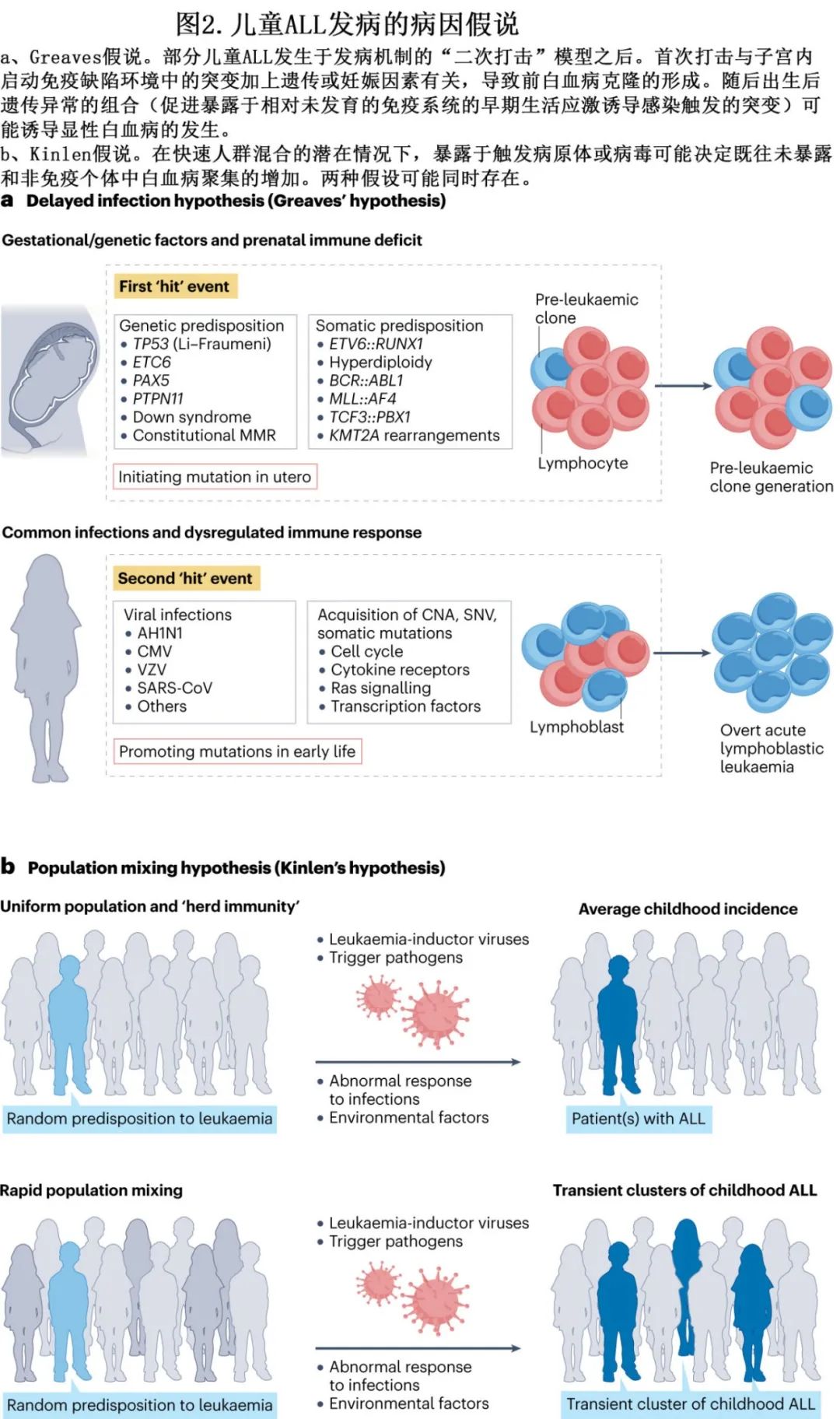
其他假说基于以下观点:婴儿期的感染模式和卫生条件可能影响下丘脑-垂体-肾上腺轴的激活或促炎性胸腺保护环境。由于反复感染导致的年龄相关性胸腺退化或 T 细胞耗竭可能反过来促进白血病前期生长。这些情况可能同时存在,并表明特定感染的直接病理作用,推测是病毒感染。但尚未明确确定得特异性促白血病因子。
遗传遗传学参与ALL的发展也可分为两类:与常见种系多态性相关的低外显率易感性(导致白血病风险增加1.5至2倍)和与罕见种系变异相关的高外显率易感性(导致相对风险显著增加>10倍)。在第一类中,除了激酶编码基因CDKN2A (9p21.3、rs3731217、rs37311249)和PIP4K2A (10p12、 rs7088318)外,全基因组关联研究(GWAS)或对复发性造血恶性肿瘤亲属的遗传分析发现,编码造血转录因子的基因存在多态性,包括ARID5B (10q21.2、rs7089424、rs10821936)、IKZF1 (7p12.2、rs4132601)、GATA3 (10p14、rs3824662)、CEBPE (14q11.2、rs2239633)、ERG (21q22.2、rs2836371)、IKZF3 (17q12、rs2290400)和ETV6 (12p13.2、rs724159945-47)。在LHPP、ELK3、BAK1和IGF2BP1基因中发现了其他易感遗传风险事件。第二类涉及与癌症易感综合征相关的罕见种系基因变异。在这一类中,有可能区分由罕见的低外显率变异引起的综合征,这些变异具有中度临床但与ALL高度似是而非的关联,以及高风险的癌症易感性变异和综合征。由低外显率变异引起的综合征通常不包括ALL或可能只优先包括ALL。第一类通常与综合征表型相关,例如Rubinstein-Taybi综合征(CREBBP、EP300)或Noonan综合征(Ras通路突变)。第二类包括体质错配修复缺陷(MLH1, MSH2, MSH6, PMS2)、Li-Fraumeni综合征(TP53)和唐氏综合征(DS)。与非DS型ALL相比,DS型ALL具有独特的生物学和遗传学特征。例如,CRFL2重排和JAK2R683突变分别发生在60%和18%的DS-ALL患者中。DS-ALL患儿的治疗相关死亡率和复发率均较高,导致预后较差。ALL也可能偶尔发生在其他特定癌症高风险的综合征中,如范可尼贫血中的T-ALL。
基于 NGS 的深度测序已鉴定出主要与家族性 ALL 有关的种系变异,如SH2B3、TP53、RUNX1、TYK2、IKZF1、PAX5和 ETV6。
机制/病理生理学
ALL是由序贯性获得的基因异常所引起,遗传亚群反映出B淋巴和T淋巴的不同分化阶段。通常来说,恶性转化影响多能干细胞祖细胞,但偶尔也涉及共同的淋巴样祖细胞。积累证据表明,ETP-ALL来源于早期T细胞前体(ETP)的恶性转化,ETP是一种未成熟胸腺细胞,从骨髓转移到胸腺,具有骨髓和淋巴细胞分化潜力和造血干细胞来源。遗传异常包括体细胞结构DNA重排、拷贝数改变和核苷酸变异,破坏细胞生长、增殖、存活和淋巴细胞分化的正常控制。它们中的大多数可决定不同的转录表达谱,概括淋巴细胞发育的模式,其他可干扰受体和非受体酪氨酸激酶信号、细胞因子介导的途径、癌基因和肿瘤抑制因子、Ras信号和染色质重塑。它们的频率和分布也取决于疾病阶段、诊断或复发,有助于获得耐药表型。
ALL的遗传学
ALL是一种遗传异质性疾病,以各种分子亚型为特征,包括非整倍体、染色体重排、拷贝数改变、序列突变和基因表达失调。这些基因改变在白血病发生中起核心作用,并与不同的基因表达特征、信号通路、临床特征、治疗反应和结局相关。预后分层确定了三个亚组:标危或低危、中危和高危,儿童的5年总生存率分别为90%、83%和62%,成人的5年总生存率分别为近50-70%、40-50%和20%,取决于风险分层、疾病亚型、诱导结束的巩固策略、儿科方案或MRD状态。
B细胞前体急性淋巴细胞白血病的遗传学
约30%的儿童 BCP-ALL 发生高超二倍体(High hyperdiploidy,51-65条染色体),但成人中仅有不到3%,并与有利结局相关。大多数高超二倍体患者存在Ras信号通路和组蛋白修饰因子的突变。染色体gain是非随机的,在子宫内早期获得,随后发生突变进化。低亚二倍体(Low hypodiploidy,32-39条染色体)在儿童 (1%) 和成人 (10%)BCP-ALL 中均表现为 TP53 突变及RB1、IKZF2和 CDKN2A/B 缺失。TP53突变是儿童病例中常见的遗传性基因事件,与Li–Fraumeni综合征相关;在成人中可能发生于单个突变的前体细胞中,称为克隆造血。近单倍体(Near-haploidy,24-31条染色体)在儿童 (2-3%) 和成人 (<1%) 中均罕见,与受体酪氨酸激酶信号转导、Ras信号转导通路 (NF1、NRAS、FLT3) 和组蛋白修饰因子 (CREBBP) 突变以及 IKZF3 失活相关。低亚二倍体和近单倍体均表现出 Ras 信号转导和磷酸肌醇3-激酶 (PI3K) 信号转导通路的激活,并与不良预后相关(表1)。
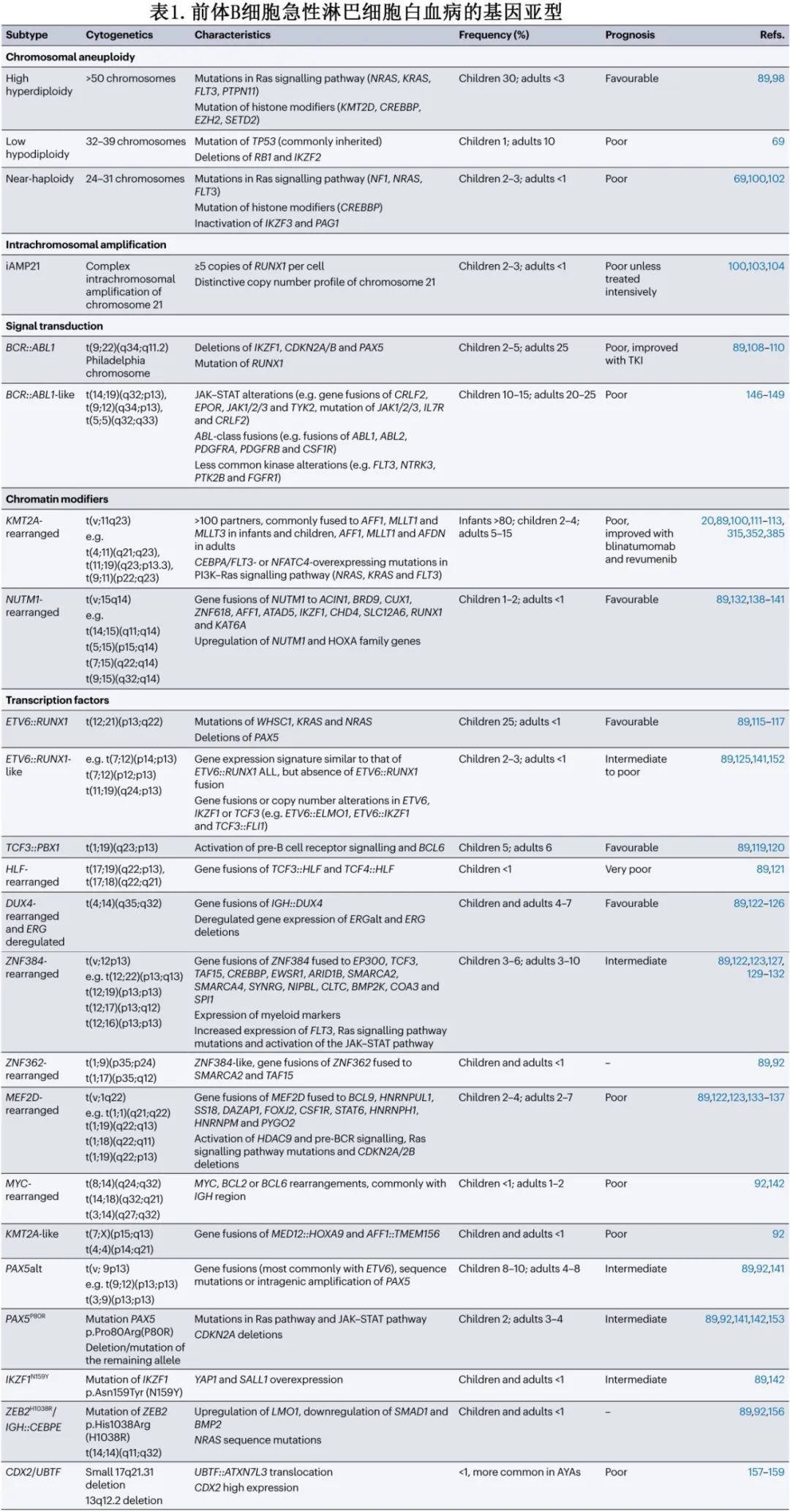
21号染色体的染色体内扩增
21号染色体的染色体内扩增 (iAMP21-ALL) 约占儿童 BCP-ALL 的3%,中位年龄为9-10岁,在成人中罕见。严重异常 iAMP21 染色体的形成源于断裂-融合-桥接循环,随后发生染色体碎裂,是拷贝数变化(插入和缺失)的复杂模式,沿着染色体或染色体片段的长度,可提供启动机制。iAMP21 染色体在患者之间具有高度异质性,最好通过其 SNP 阵列或全基因组测序 (WGS) 的特征性拷贝数谱进行鉴定。一系列特征性的继发性改变与 iAMP21-ALL 相关,其中最常改变的通路为iAMP21-ALL 中的JAK–STAT信号转导、Ras信号转导、B细胞分化和其他转录调节因子。该亚型最初与不良预后相关,但强化治疗显著改善了其结局,尤其是在儿童中(表1)。
染色体重排
易位t(9;22)(q34;q11.2)(Ph) 是成人中最常见的重排,与BCR::ABL1融合的表达相关,其编码导致白血病发生的酪氨酸激酶信号激活蛋白。Ph 阳性 BCP-ALL 的频率随年龄增长而增加,儿童为5%,成人为25%。酪氨酸激酶抑制剂 (TKI) 治疗显著改善了这些患者的预后。
KMT2A(混合谱系白血病;MLL)重排常见于婴儿(0-12个月)ALL(>80%),成人患者中约有15%,与不良预后相关。婴儿KMT2A重排ALL的高发病率在一岁时达到高峰,在儿童和青年阶段下降,然后逐渐上升,直到55岁。KMT2A有多个易位伴侣(>100),在婴幼儿和儿科ALL患者中通常与AFF1、MLLT1或MLLT3融合,而在成人ALL患者中则经常与AFF1、MLLT1或AFDN融合。KMT2A重排根据其表达谱可分为CEBPA/FLT3或NFATC4过表达亚组,该亚型表现出HOX簇基因的过表达和激酶-PI3K–Ras信号通路的激活突变。
导致ETV6:::RUNX1(TEL::AML1) 融合的t(12;21)(p13;q22) 易位是儿童 BCP-ALL 中最常见的结构改变 (25%),在成人中则罕见 (<1%),并且在所有年龄组中均与良好预后相关。ETV6::RUNX1融合通常在子宫内获得,需要额外的基因改变来诱导显性白血病,如WHSC1、KRAS和 NRAS 突变,以及 PAX5 缺失。导致TCF3::PBX1融合的t(1;19)(q23;p13) 易位存在于约6%的儿童和成人 BCP-ALL 患者中,与当前化疗方案的有利结局相关;TCF3::PBX1融合基因可激活前 B 细胞受体信号转导(前BCR)并诱导 BCL6 上调(表1及下图)。相比之下,<1%的 BCP-ALL 中存在TCF3::HLF和TCF4::HLF融合基因,且与不良预后相关。Notch 信号通路的激活已在 HLF 重排的 ALL 中得到证实。利用转录组测序已发现多种新的基因改变,而常规分析未鉴定出来。约7%的 BCP-ALL存在DUX4 重排,通常与极佳的预后相关。双同源盒转录因子基因DUX4插入IGH 位点导致 DUX4过表达,DUX4 重排通常与 ERG 的基因内缺失相关,导致新 ERG 亚型 ERGalt 的表达和/或 ERG 缺失。已发现 DUX4 重排 ALL 的两个不同基因表达簇,一个以 ERG/TBL1XR1 改变和 NFATC4 高表达为特征,而另一个与NRAS、IKZF1和 KMT2D 突变以及 CEBPA/FLT3 高表达相关。
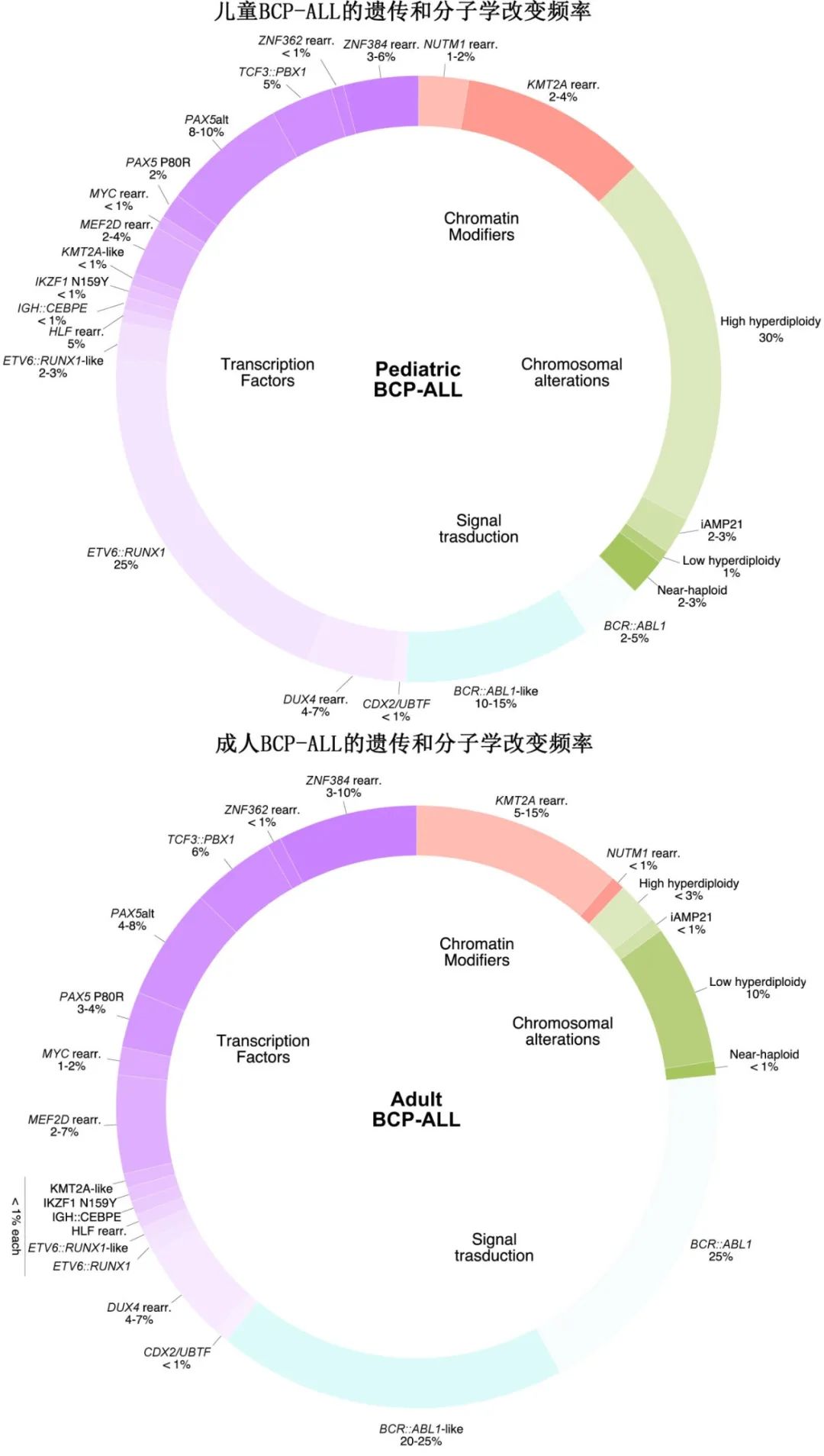
ZNF384 重排 ALL 的特征为独特的免疫表型,CD10弱表达,髓系标志物 CD13 和/或 CD33 表达异常。其占儿童BCP-ALL的3-6%,占成人BCP-ALL的3-10%,预后中等。ZNF384 重排约占儿童 B/髓系混合表型急性白血病 (MPAL) 的50%。在 ALL 中发现了超过10种不同的 ZNF384 融合伴侣(最常见的是EP300、TCF3、TAF15)。与FLT3 上调、Ras信号通路突变和JAK–STAT通路激活相关。在 ZNF384 样 ALL 中还观察到 ZNF362 重排(SMARCA2::ZNF362和TAF15::ZNF362)(基因表达谱与 ZNF384 融合相似)(儿童和成人中均<1%)。
复发性MEF2D融合在儿童BCP-ALL中发生率为2-4%,成人BCP-ALL中发生率为2-7%,已确定为预后不良亚型。有10个基因为MEF2D的融合伴侣(表1),最常见的为BCL9和HNRNPUL1,占病例数的80%以上。MEF2D重排的特征为HDAC9表达增加、CDKN2A/2B缺失、编码前B细胞受体信号的基因上调、Ras信号通路突变以及对组蛋白去乙酰化酶(HDAC)抑制剂的敏感性。
约1%的儿童 BCP-ALL 携带NUTM1 重排,尤其是在无 KMT2A 重排的婴儿,以及<1%的成人 BCP-ALL 中。该亚型的特征为 NUTM1 与各种基因对应物融合、NUTM1和 HOXA 基因家族上调、Notch信号通路激活和预后良好。
BCL2、MYC或 BCL6 重排(通常伴有IGH)在 BCP-ALL 成人中的发生率约为2%,但在儿童中很少发生 (<1%),并导致重排等位基因的过度表达和白血病原始细胞增殖的激活。这些易位与早期治疗失败相关(表1)。
拟表型亚型(Phenocopy subtypes)
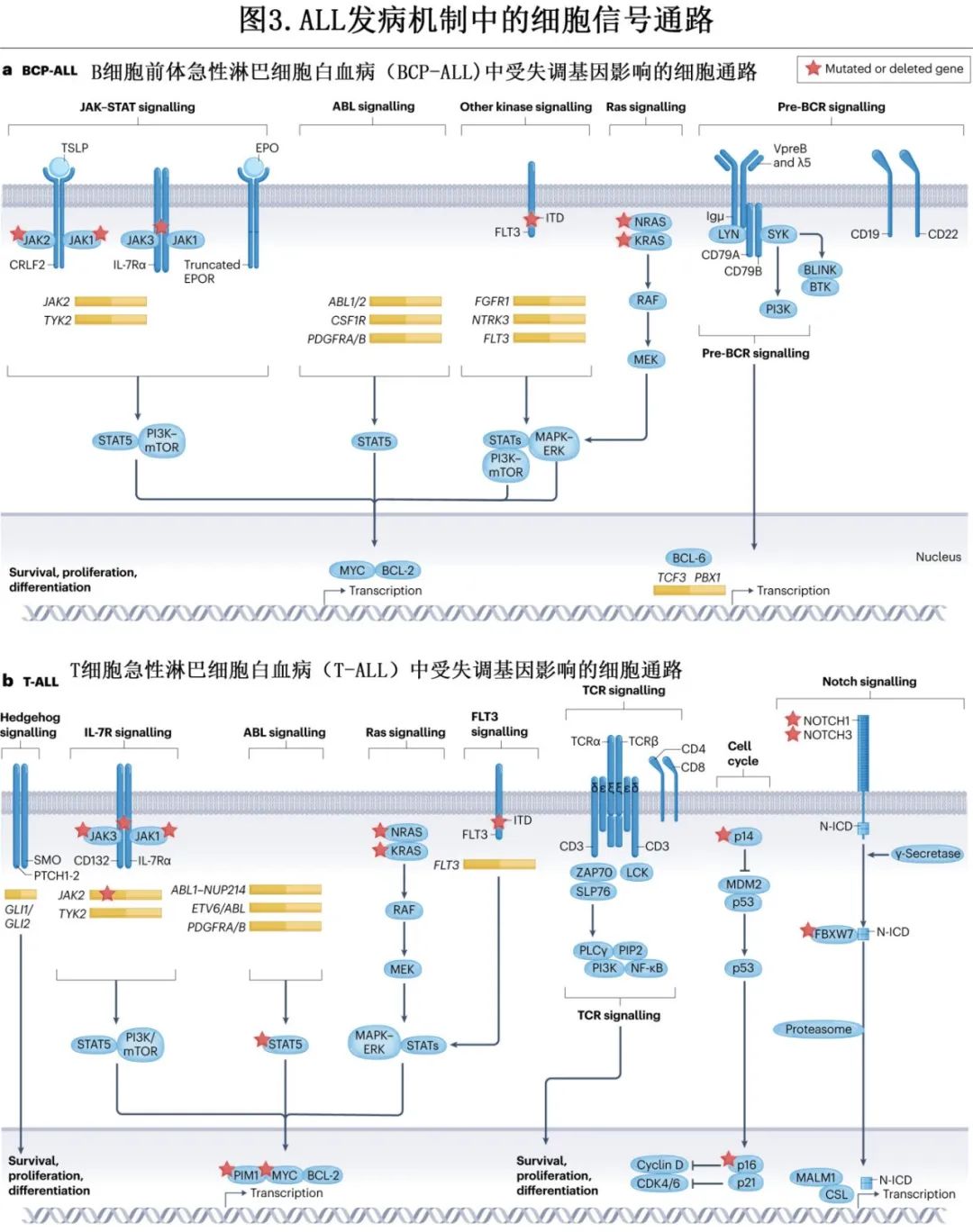
BCR::ABL1样ALL的特征在于转录谱与Ph阳性ALL相似,但缺乏BCR::ABL1融合基因,其占儿童BCP-ALL 的的10-15%,占成人 BCP-ALL 的20-25%。该亚型具有参与酪氨酸激酶信号转导和细胞因子受体的多种基因改变,包括JAK–STAT改变(例如CRLF2、EPOR、JAK1/2/3和 TYK2 基因融合,JAK1/2/3、IL7R和 CRLF2 基因突变)、ABL类融合(例如ABL1、ABL2、PDGFRA、PDGFRB 和CSF1R)和其他不太常见的激酶改变(例如FLT3、NTRK3、PTK2B和 FGFR1 中)(图3a及上图)。Ph 样 ALL 与不良预后相关,但 TKI 联合化疗治疗 ABL 类或其他不太常见的融合(如TRK 抑制剂治疗 NTRK3 融合阳性白血病中)可获得更好结局。
ETV6::RUNX1样 ALL 具有与ETV6::RUNX1 ALL相似的基因表达特征和免疫表型,但不存在ETV6::RUNX1融合。ETV6::RUNX1样 ALL 表现出ETV6、IKZF1或 TCF3 的基因融合或拷贝数改变,导致淋巴细胞发育失调。该亚型在儿童期(高达3%)比成人 (< 1%)BCP-ALL更常见,预后中等。KMT2A 样亚型在 BCP-ALL 中罕见 (<1%)。MED12::HOXA9和AFF1:: TMEM156 融合也在 KMT2A 样病例中有所发现。
其他亚型
已经定义两种 BCP-ALL 亚型,其具有不同的、PAX5 驱动的改变:PAX5改变 (PAX5alt) BCP-ALL 包括 PAX5 重排(最常见于ETV6)、序列突变或基因内扩增,占儿童 BCP-ALL 的8-10%和成人 BCP-ALL 的4-8%;PAX5P80R BCP-ALL 的特征为热点突变 PAX5P80R 和其余等位基因缺失或突变,发生在约2%的儿童和3%的成人 BCP-ALL 中。PAX5P80R BCP-ALL 涉及 Ras 和JAK–STAT通路突变、CDKN2A缺失、PI3K–AKT–雷帕霉素机制靶点 (mTOR) 信号转导上调和细胞粘附分子下调。在成人 PAX5P80R中,这些亚型与中等预后和良好预后相关。
高危 BCP-ALL 中存在不同的 IKZF1 改变,最常见的是基因内 IKZF1 缺失。IKZF1N159Y BCP-ALL的点突变是一种具有独特转录谱的新亚型(在 BCP-ALL 中< 1%),可导致 YAP1 和 SALL1 过表达。
<1%的病例携带ZEB2H1038R和IGH::CEBPE融合的点突变,与LMO1上调、SMAD1和 BMP2 下调以及 NRAS 序列突变相关。携带 ZEB2 突变的 BCP-ALL 与无事件生存期 (EFS) 差和复发率高相关(表1)。
CDX2/UBTF ALL 表现为独特的基因表达特征和独特的双基因组改变,即17q21.31的 UBTF 局灶性缺失导致UBTF:ATXN7L3融合,13q12.2的 FLT3 和 PAN3 基因缺失导致 CDX2 高表达。CDX2/UBTF ALL 在 BCP-ALL 中罕见 (<1%),但在青少年和年轻成人(AYA;15-40岁,根据大多数报道的定义)患者中更常见 (1.4%),富集于女性(男女比例0.2),由于首次诱导疗程失败风险高和复发率高,预后较差。
T细胞急性淋巴细胞白血病的遗传学
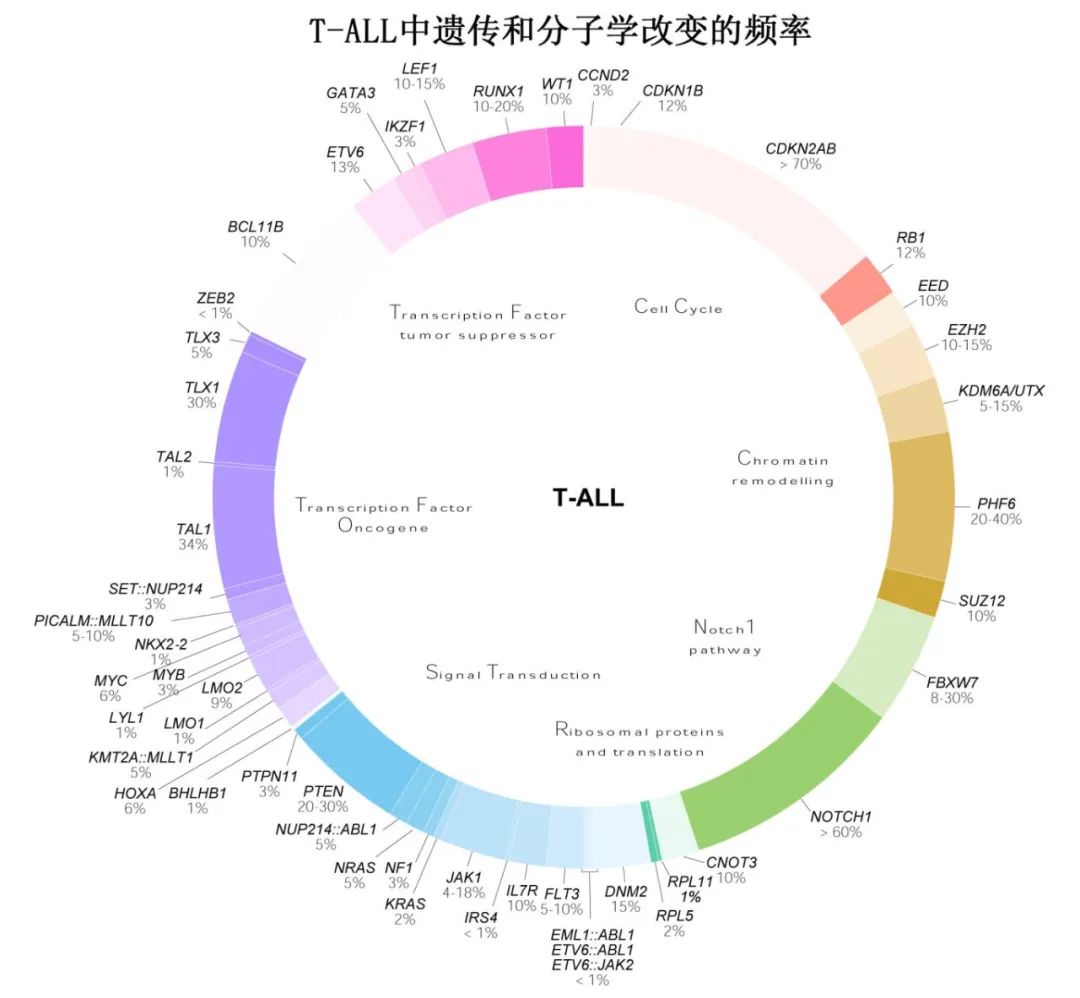
独特的基因表达特征可确定 T-ALL 内的特定分子亚组,并与形态细胞化学和流式细胞术诊断标准整合。T-ALL的特征为过表达转录因子基因,后者与非重叠基因病变(突变或易位)共存。因此,ICC提出了8个亚组作为临时亚型(HOXA失调、SPI1、TLX1、TLX3、NKX2、TAL1-2、LMO1-2重排 T-ALL和T-ALL伴涉及其他螺旋-环-螺旋家族成员[包括 LYL1 或OLIG2/BHLHB1]的重排)(表2和下图)。尽管不断努力,但在定义不同的 T-ALL 亚型方面仍缺乏共识,部分原因在于难以检测驱动癌基因失调的常见非编码基因组改变。LYL1、LMO2、SPI1、MEF2C和 HOXA 家族基因,以及SET::NUP214和NUP98 融合的表达升高,均与 LYL1/LMO2 失调亚型相关。已经确定了一种以 GATA3 突变为特征的新亚型,即GATA3R276Q,它影响 zebrafish中的淋巴细胞发育。SPI1重排亚型与TCF7::SPI1和STMN1::SPI1融合相关,使 SPI1 表达增加,预后不良。HOXA 失调的亚型主要涉及 KMT2A 重排、MLLT10重排、HOXA10重排和 HOXA 家族基因的过表达。LYL1/LMO2/SPI1/HOXA 过表达患者表现出 ETP-ALL 或近 ETP 免疫表型。TLX3 和 TLX1 失调 ALL以 TLX3 或 TLX1 重排为特征,可在20-30%的病例中观察到。TLX3 和 TLX1 通常分别与 BCL11B 或 TRA/B 融合,并显示 TLX3 或 TLX1 的过表达。NKX2.1失调的亚型与NKX2.1重排和过表达相关。
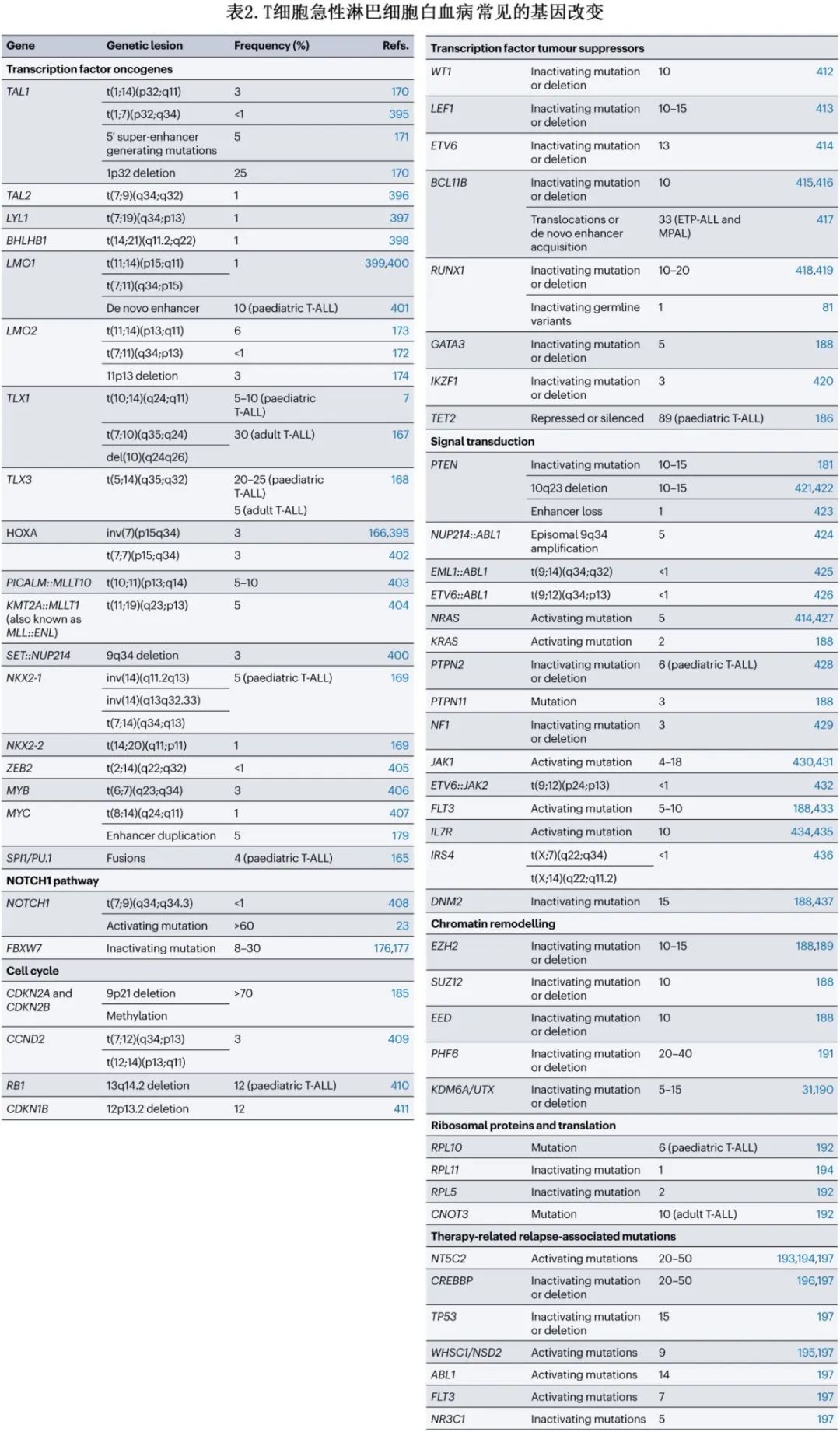
遗传和免疫表型特征之间存在典型相关性,包括:表达早期胸腺细胞抗原 (CD5+) 的白血病中存在HOXA 或 TLX3 失调;存在成熟 (sCD3) 或皮质 (CD1a+) 淋巴细胞特征时的TAL/LMO 失调;表达皮质抗原 (CD1a+) 的白血病中存在 TLX1/TLX3 或NKX2.1/2.2失调。遗传异质性和低危的未成熟 ETP 表型主要为 HOXA 失调,较少属于T/髓系 MPAL 型,可通过 BCL11B 活化予以特异性鉴别。
以 TLX1/TLX3 过表达为特征的 T-ALL 患者的白血病原始细胞可在中期 (CD1a+CD4+CD8+) 被阻断,并发生皮质或皮质后免疫表型。TAL1/LMO1 失调亚型的特征为多种融合,包括STIL::TAL1、TAL2、LMO2和 LMO1 融合,以及 TAL1 和 LMO1 过表达。有趣的是,在20-30%的 T-ALL 中,TAL1过表达可继发于新生增强子的产生。在10-15%的病例中观察到 LMO1 或 LMO2 的基因改变。具有更成熟、皮质、皮质后或晚期成熟 T 细胞免疫表型 (CD3+CD4+CD8+) 的 T-ALL 改变通常显示NKX2.1、TAL1和 LMO1 基因活化。
参与T-ALL的体细胞突变基因可分为7个功能性分类:转录因子癌基因;细胞周期调节剂;肿瘤抑制转录因子;信号通路;染色质重塑通路;核糖体蛋白和蛋白质翻译;治疗相关突变(表2)。
NOTCH1 是 T-ALL 中最常发生突变的转录因子基因(>60%),其次为泛素连接酶基因FBXW7(其也可导致 NOTCH 通路过度活化)、PHF6和PTEN。NOTCH1 通路在多个 T 细胞进程中具有关键作用,包括干细胞自我更新、增殖和胸腺分化。因此,其失调或下游通路改变(如 MYC 信号或PTEN–AKT信号)可导致白血病转化或代谢脱轨(metabolic derailment)(图3b)。因此,靶向 NOTCH1 是该领域研究的一个活跃领域。在作为 T-ALL 突变靶点的细胞周期调控因子中,CDKN2A/B最为突出。事实上,这些肿瘤抑制基因的缺失是 T-ALL 中最常见的基因异常(>70%的患者),但其他细胞周期调节基因如CCND2、RB1和 CDKN1B 在 T-ALL 中也可发生复发性突变。在相当多的T-ALL 病例 (>10%) 中,多种肿瘤抑制转录因子也可表现出基因异常,包括WT1、LEF1、ETV6、BCL11B和RUNX1(表2)。有趣的是,尽管 TET2 在 T-ALL 中很少发生突变 (<1%),但一项研究报道,其在 T-ALL 中可经由启动子超甲基化非常频繁地被抑制或完全沉默(>80%的病例),从而揭示了用阿扎胞苷治疗这些患者的治疗机会。在信号通路的靶基因中,包括JAK1、FLT3、IL7R、DNM2和 PTEN 的基因存在相对常见的突变(表2)。重要的是,PTEN功能缺失突变与 NOTCH1 抑制和糖皮质激素治疗抵抗有关。在过去的十年中,另一类越来越受关注的领域为染色质重塑基因的改变,如多梳抑制复合体2 (PRC2)成员、SUZ12、EED和EZH2突变,或组蛋白去甲基化酶基因KDM6A(也称为UTX)突变。此外,最常见的突变染色质重塑因子为X 连锁基因PHF6(20-40%的病例,多为男性)。与儿童 T-ALL 相比,在成人 T-ALL 中更常发现与表观遗传调节因子(IDH2、DNMT3A、CHD4、ASXL1、CREBBP和EZH2)、JAK–STAT信号转导(JAK3和JAK1)和 Ras 信号转导 (NRAS) 相关的突变基因。
最后,核糖体蛋白或翻译基因存在常见突变,包括突变 RPL3 和CNOT3。最后,值得一提的是另一类与治疗相关和复发相关的突变,包括驱动对6-巯基嘌呤耐药的 NT5C2 突变或驱动对糖皮质激素耐药的 CREBBP 突变(以及较少见的 NSD1 和 NR3C1 突变)(表2)。
参考文献
Pagliaro, L., Chen, SJ., Herranz, D. et al. Acute lymphoblastic leukaemia. Nat Rev Dis Primers 10, 41 (2024). https://doi.org/10.1038/s41572-024-00525-x

