打进体内的局麻药都去哪儿了?
时间:2024-07-23 14:04:05 热度:37.1℃ 作者:网络
想要搞清楚这个问题,就要熟知局麻药的药代动力学。
局麻药最常被注射到靠近目标部位的血管外组织。其血药浓度受所用局麻药的总剂量、全身吸收程度、组织重新分布和消除速率的影响。患者特异性因素如年龄、心血管和肝功能以及血浆蛋白结合率等也会影响随后的血药浓度。了解上述因素,可将局麻药的临床应用范围最大化,同时将其全身毒性相关潜在并发症降至最低。
1. 全身吸收
局麻药全身吸收的速率和程度受多个因素影响,包括药物总剂量、给药部位、局麻药特有的理化性质和血管收缩药(肾上腺素)的使用。
(1)局麻药剂量:对于任何给药部位,全身吸收程度和药峰浓度(Cmax)与局麻药的总剂量呈正相关。
此外,增加吸收速率也会降低达峰时间(Tmax)。在常用临床剂量的范围内,量效关系接近线性,并且相对不受麻醉药浓度或注射速度的影响。
(2)组织灌注:神经周围组织的灌注程度显著影响全身吸收,所以在高灌注的神经周围组织中使用局麻药会导致更高的Cmax和更短的Tmaxo因此,全身吸收率从高到低依次是胸膜腔内>肋间>骶尾部>硬膜外>臂丛>坐骨/股骨>皮下组织。
(3)理化性质:全身吸收的速率也受局麻药特有的理化性质影响。通常,脂溶性强的局麻药会使全身吸收降低。其脂溶性越大,越容易被富含脂质的轴突膜和神经周围组织隔离。
(4)血管收缩药:肾上腺素抵消了大多数局麻药固有的血管舒张特性。对于脂溶性较低的局麻药而言,肾上腺素可显著降低其Cmax,而随着脂溶性的增强,增加神经和周围组织的结合成为增加局麻药全身吸收率的关键因素。
(5)全麻的吸入麻醉药:动物数据表明,在吸入麻醉药的全身麻醉下给予局麻药时,血药浓度显著增加。
2.分布
全身吸收后,局麻药迅速分布于全身组织中。分布模式(和相对组织浓度)受灌注、分配系数和特有组织区域数量的影响。高灌注器官(脑、肺、心脏、肝脏和肾脏)负责最初的快速摄取(α阶段),然后是低灌注的组织(肌肉和肠道)负责较慢的重新分配(β阶段)。由于局麻药在肺组织被迅速提取,其血药浓度在通过肺血管时显著降低。
3.生物转化和消除
化学结构决定了局麻药的生物转化和消除(图5-4)。
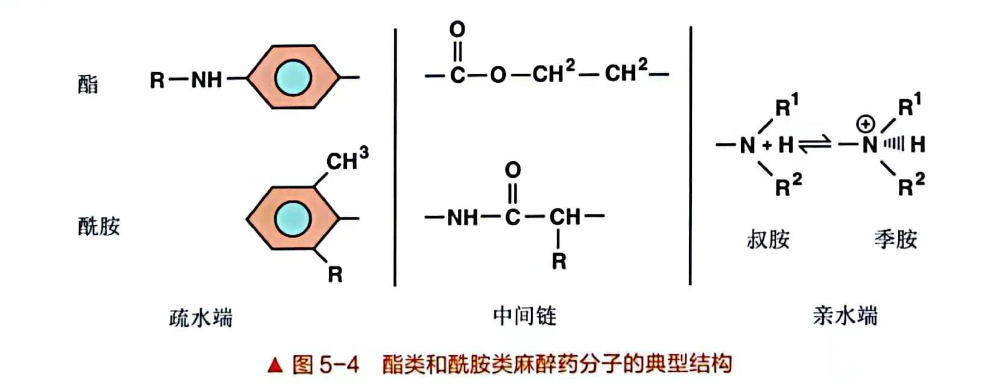
氨基酰胺在肝脏中通过细胞色素P450酶经由N-去烷基化和羟基化代谢。氨基酰胺代谢高度依赖于肝灌注、肝提取和酶功能。因此,由于肝硬化和充血性心力衰竭等症状,局麻药清除率降低。氨基酰胺的代谢产物通过肾脏排泄,<5%的非代谢性局麻药由肾脏排泄。
氨基酯类局麻药迅速被血浆胆碱酯酶代谢。普鲁卡因和苯佐卡因代谢生成对氨基苯甲酸(PABA),使用这些局麻药出现罕见的过敏反应与它有关。基因异常的血浆胆碱酯酶或服用胆碱酯酶抑制药的患者氨基酯类代谢减少。理论上讲,这些会增加全身毒性作用的风险,但临床证据不足。
4.临床药代动力学
局麻药的代谢具有重要的临床意义,因为全身毒性(主要由Cnax决定)取决于全身吸收和消除之间的平衡。
(1)血浆蛋白结合:局麻药主要与组织和血浆蛋白结合,但全身毒性与游离(未结合)血浆浓度有关。因此,局麻药与血浆蛋白的结合降低了体循环中的游离血浆浓度,并且降低了全身毒性的风险。血浆蛋白结合的程度主要取决于血浆中α1-酸性糖蛋白和白蛋白的水平,也受血浆pH的影响。
降低血浆蛋白(肝硬化、妊娠和新生儿)的临床条件降低了其结合能力。此外,随着pH降低,蛋白质结合的百分比降低。因此,在酸中毒(癫痫发作、心脏骤停和肾衰竭)的情况下,未结合药物的量增加。
(2)肝清除率:肝清除率的改变也可能影响局麻药的消除。例如,新生儿的肝微粒体酶发育不全,导致氨基酰胺局麻药的消除减少。一些药物如β受体阻滞药、H2受体拮抗药和氟伏沙明可抑制特定的肝微粒体酶,也可能有助于减少氨酰胺局麻药的代谢。肝血流量减少会导致局麻药血药浓度大幅度增加。
所有,以上描述的影响局麻药全身吸收、分布和患者特异性的因素都应该考虑,以尽量减少全身毒性的风险。这些因素构成了当前局麻药“最大剂量”推荐的基础。
降低血浆蛋白和血浆蛋白结合程度,以及降低肝血流量(例如充血性心力衰竭患者)等临床条件,会增加局麻药全身毒性(LAST) 的风险。

