Adv Sci:上海科技大学谢成英团队揭示KRAS突变癌症中转录暂停释放和补体激活中的CDK9/PP2A/ERK网络
时间:2024-09-13 06:01:02 热度:37.1℃ 作者:网络
选择性抑制转录延长因子(P-TEFb)复合物代表了一种有前途的癌症治疗方法,但CDK9抑制剂(CDK9i)目前主要限于某些血液系统恶性肿瘤。
2024年9月10日,上海科技大学谢成英独立通讯在Advanced Science 在线发表题为“Unraveling the CDK9/PP2A/ERK Network in Transcriptional Pause Release and Complement Activation in KRAS-mutant Cancers”的研究论文。该研究虽然在体外观察到了各种KRAS突变癌症类型对CDK9靶向疗法的初始反应,但它们在裸鼠异种移植模型中的疗效远不能令人满意。
从机制上讲,CDK9抑制会导致ERK-MYC信号的补偿性激活,同时伴有原癌基因的恢复、立即早期基因(IEG)的上调、补体C1r-C3-C3a级联的刺激以及肿瘤免疫抑制的诱导。涉及CDK9/Src相互作用的PP2Ac活性的“矛盾”调节有助于ERK磷酸化和RNA聚合酶II(PolII)的暂停释放。CDK9和KRAS/MAPK信号通路的共同靶向消除了ERK-MYC激活并阻止了受体酪氨酸激酶介导的反馈激活,从而更有效地控制KRAS突变癌症并克服KRASi耐药性。此外,通过补体系统干预调节肿瘤微环境(TME)可增强对CDK9i的反应并有效抑制肿瘤生长。总体而言,临床前研究为开展临床试验建立了一个强大的框架,该试验采用KRASi/SOS1i/MEKi或免疫调节剂与CDK9i结合,同时靶向癌细胞及其与TME的串扰,从而改善KRAS突变患者的反应。

自从发现编码蛋白激酶的致癌基因以来,人们对激酶抑制作为一种靶向治疗策略的乐观态度现已转化为临床现实,包括细胞周期依赖性激酶(CDK),特别是已获批准的CDK4/6抑制剂和正在进行的其他CDK临床试验。其中,正转录延伸因子b(P-TEFb)的催化亚基CDK9促进RNA聚合酶II(PolII)的暂停释放并加强信号反应基因的转录延伸。急性抑制CDK9会导致转录的短暂抑制和Mcl-1和c-Myc等短寿命蛋白的优先消耗。
因此,CDK9抑制剂(CDK9i)已在Mcl-1或MYC驱动的恶性肿瘤的临床前模型中显示出治疗效果,从而推动其进入临床试验。尽管CDK9i在体外研究中显示出对实体瘤细胞的强效细胞毒性,但在体内模型中,内在抗性占主导地位。由于效力和特异性问题,美国食品药品监督管理局(FDA)的批准仍然难以实现。疗效不令人满意意味着对CDK9相互作用的大分子复合物、CDK9i激活的致癌途径、复杂而冗余的转录机制或其他药物驱动的补偿机制的理解不完整。因此,阐明CDK9抑制在实体瘤中引发的细胞补偿可能会显著增强这种强效干预对高度难治性癌症的有效性。
KirstenRas(KRAS)突变是人类癌症中最常见的变异之一,发生在约25%的患者中。这些频率在固有致命性恶性肿瘤中尤其升高,例如胰腺导管腺癌(PDAC)、结肠腺癌(COAD)和非小细胞肺癌(NSCLC)。在氨基酸G12、G13和Q61处具有明显突变的KRAS与肿瘤侵袭性和不良的患者预后有关。针对KRASG12C变体的特异性抑制剂的成功开发标志着KRAS靶向治疗领域的显著进步。然而,这些药物的治疗潜力受到内在或迅速出现的获得性耐药性的阻碍,并且缺乏针对其他KRAS突变体的可用药物。此外,KRAS突变癌症的异质性导致KRAS抑制剂(KRASi)作为单一疗法的疗效有限;而与其他靶向药物,包括SHP2、EGFR、PD-L1、CDK4/6、PLK1和IRE1α联合使用,可显著增强抗肿瘤活性并克服耐药性。因此,深入研究KRAS抑制剂的耐药机制并阐明可用于联合治疗的新型分子靶点对于大多数携带不同KRAS突变的患者至关重要。
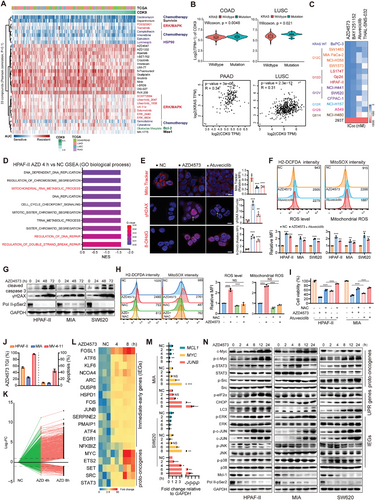
CDK9抑制会导致KRAS突变癌细胞中的氧化应激增加和线粒体功能障碍。(图源自Advanced Science )
对KRASi的反应与治疗期间肿瘤微环境(TME)重塑有关,其特征是效应T细胞浸润增加、抗肿瘤M1巨噬细胞复极化和自然杀伤(NK)细胞募集,这增加了KRAS抑制后对免疫疗法的敏感性。将KRASi与免疫检查点阻断(ICB)相结合的协同效益仅在免疫原性肿瘤模型中观察到。然而,在临床环境中实现这种协同作用已被证明是一项艰巨的挑战,因为它无法增强对免疫原性冷性肿瘤(如PDAC)的疗效。此外,对KRASi表现出抗性的恶性肿瘤通常伴有免疫抑制性TME,并对ICB治疗产生交叉抗性。这凸显了探索靶向药物的免疫调节特性和其他免疫成分的治疗前景的迫切必要性。
在本研究中,作者采用转录组测序和相互作用组筛选来确定赋予肿瘤对CDK9i耐受性的因素。作者发现了涉及蛋白激酶和磷酸酶的关键机制转换,它们动态调节PolII和ERK依赖的转录延长。此外,作者证明了CDK9i在KRAS突变癌症中的体内疗效有限,这不仅可以归因于肿瘤细胞中激活的促生存信号,还可以归因于补体系统协调的免疫抑制TME。作者的研究结果有望建立一种致癌基因驱动的组合治疗策略,以增强CDK9i和KRAS信号抑制剂的抗肿瘤活性,同时减轻免疫抑制并克服耐药性。
参考消息:
https://onlinelibrary.wiley.com/doi/10.1002/advs.202404926

