双向压力:呼吸机-肺-肾交互作用的综述
时间:2024-09-14 18:02:47 热度:37.1℃ 作者:网络
急性肾损伤和需要机械通气的呼吸衰竭都是危重症的常见并发症。这两个器官系统中的任何一个出现损伤都会增加另外一个出现损伤的概率。因此,急性肾损伤合并需要机械通气的患者的发生率很高,这严重影响了重症监护室的结局(包括死亡)率。尽管我们对呼吸机-肺-肾交互作用的机制进行了数十年的研究,但仍然存在一些未知的知识,目前的治疗策略以脏器支持为主。在本文中,我们概述了目前机械通气导致急性肾损伤机制的认识,包括:1)机械通气对肾灌注的影响,2)正压通气对神经激素通路的激活,3)呼吸机诱导的肺损伤时释放的炎症介质的作用。另外也对急性肾损伤增加呼吸衰竭风险的机制进行了综述。接下来概述了目前预防危重症患者肺-肾损伤的治疗方法,包括液体和血管加压药的管理、呼吸机策略和急性肾损伤的治疗。最后讨论部分,我们概述了一些可能为新的治疗方法提供理论基础的新研究。
机械通气是急性肾损伤(AKI)发展的独立危险因素。相反,AKI也是呼吸衰竭需要机械通气的独立危险因素。大约75%的AKI患者在重症监护室(ICU)住院期间将同时接受机械通气。与单独的AKI或呼吸衰竭相比,二者皆有的后果很危险,死亡率增加了4-6倍。此外,机械通气期间的AKI也与住院时间延长、ICU住院时间增加和呼吸机使用天数增加有关。
临床前和临床研究将肺和肾之间的细胞、分子和力学交互作用描述为协同效应对死亡率影响的机制。目前治疗仍然是以器官支持为主,仍然存在一些知识盲区阻碍新的治疗方法的发展。关于机械通气对肾脏生理的影响以及肾功能改变与肾实质损伤之间的关系,仍缺乏数据。参与肺肾之间相互作用的确切炎症介质也不清楚。在这篇综述中,描述了目前对呼吸机-肺-肾交互作用机制的理解。我们还对目前在机械通气和AKI期间预防肺损伤和肾损伤的治疗方法进行了批判性的回顾。最后,我们回顾了一些目前的知识盲区和新的研究机会,可能会促进新的治疗策略的发展,从而挽救生命。
机械通气导致AKI的机制
2013年,Van den Akker等人进行了一项meta分析综述,其中包括31项研究,主要关注机械通气的使用与随后出现AKI之间的关系。研究表明,有创机械通气独立地将AKI的发生率增加了3倍。最近一项更新的研究显示,发现机械通气期间AKI的发生率仍然高达39%。研究还发现,大多数AKI病例发生在机械通气开始后的1-3天,这表明了潜在的因果关系。在这部分内容,我们回顾了一些目前机械通气影响肾功能并可能导致AKI的机制(图1)。
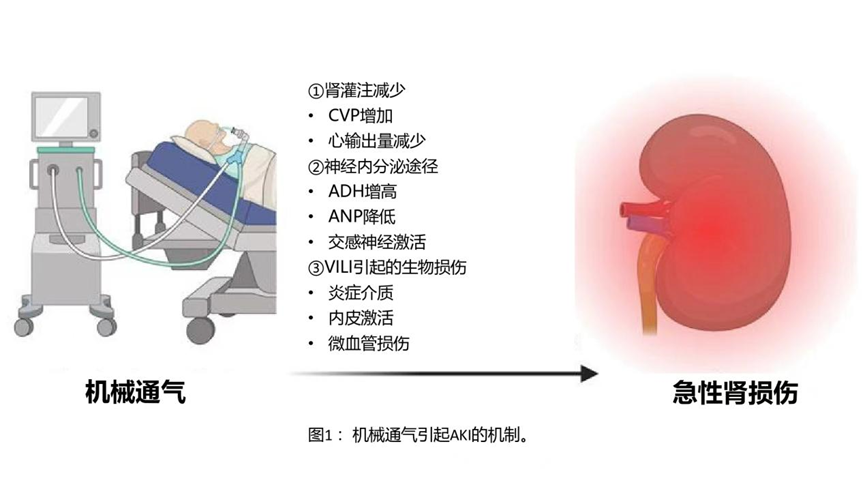
解释机械通气期间AKI高发病率的最佳机制是正压通气对全身血流动力学影响。75年前,Drury等人证明,在健康志愿者中,持续气道正压(CPAP)水平的增加与尿素清除率呈负相关。为了解释这些发现,作者提出,机械通气期间胸腔内压的增加会导致静脉回流和心输出量减少,从而导致肾灌注的减少。这一假设在随后的犬类研究中得到了证实,CPAP或呼气末正压(PEEP)水平的增加与肾灌注的减少相关。此外,通过用药提高心输出量可以恢复肾血流量。除了对心输出量的影响外,正压通气还通过增加中心静脉压(CVP)和静脉充血来影响肾灌注。肾灌注压等于平均动脉压(MAP)–CVP。因此,当CVP随着正压通气而增加时,MAP恒定,肾灌注压将降低。受试者CVP和PEEP的增加均与肾小球滤过率(GFR)的降低相关。
正压通气激活的神经激素通路也与机械通气过程中发生的肾功能变化有关。Fewell和Bond是最早揭示这些机制的人之一,他们证明了在正压通气之前切断肾神经可以改善尿量和GFR。在随后的研究中,在人类和动物模型中都发现了肾素-血管紧张素系统(RAS)的激活、抗利尿激素(ADH)的释放增加(也称为血管加压素)以及心钠肽(ANP)的减少。这些途径除了促进钠水潴留,还通过肾前血管收缩进一步降低肾血流量和肾小球滤过率。尽管胸腔内压力对心房牵张感受器、颈动脉压力感受器和交感系统激活的影响都可能起作用,但有助于激活这些神经体液途径的确切机制尚未完全阐明。
最后,发现在高潮气量和高气道压的损伤性机械通气过程中,损伤的肺和肾之间的炎症相互作用会导致肾小管损伤和细胞凋亡。在一项具有里程碑意义的研究中,Imai等人的研究表明,因高潮气量通气而发生呼吸机诱导肺损伤(VILI)的兔的血清,在健康兔的体外和体内均可引起肾小管细胞凋亡。这些发现提供了肺损伤产生的全身炎症介质与下游肾脏后果(即生物创伤)之间的直接联系。我们小组和其他人的后续研究发现,VILI会导致内皮炎症和肾脏微血管功能障碍,VILI对肾脏的影响仍然是一个活跃的研究领域。
AKI致肺损伤机制的研究进展
呼吸衰竭是导致AKI患者死亡的主要原因之一,特别是在需要机械通气的情况下。我们最近发现,机械通气期间的AKI与气体交换受损有关,可以通过动脉血中氧分压与吸入氧浓度的比值来测量。还发现AKI与呼吸力学受损有关,与没有AKI的机械通气患者相比,AKI患者的肺顺应性降低,驱动压增加,平台压增加。少尿患者的容量超负荷肯定有助于这些发现,但我们发现,即便是在总液体平衡为负的患者中,AKI对呼吸力学和气体交换的影响仍然显著。这些结果表明,也可能跟其他机制如从肾脏到肺的炎症交互作用有关。在这部分内容,我们回顾了AKI致肺损伤的一些炎症介质,这些介质可能是可以改变的(图2)。
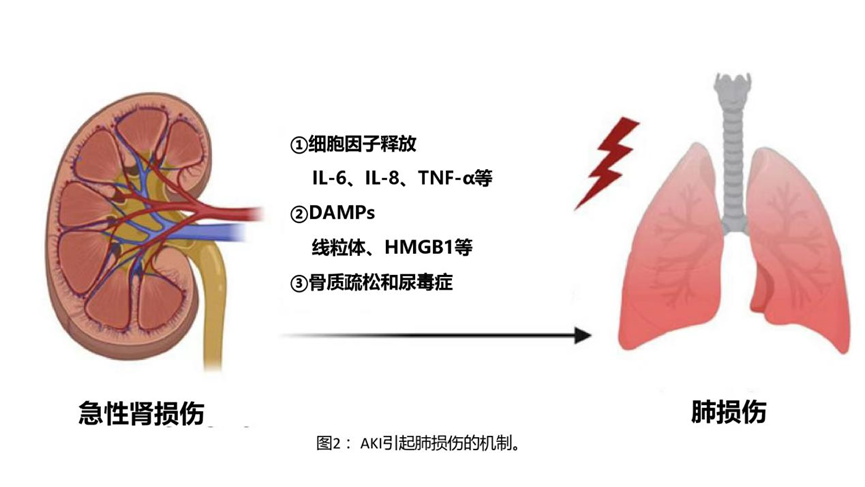
早在20世纪50年代就已经描述了AKI引起的肺损伤,最初认为是由于肾功能下降导致尿毒症毒素的积累。Yabuuchi等人近期的研究表明,AKI发生后尿毒症毒素吲哚硫酸盐在肺内积聚,导致水通道蛋白5表达降低。肺泡上皮细胞利用水通道蛋白5清除肺泡腔的水分,因此水通道蛋白的下调可能是AKI所致肺水肿的重要机制,尤其是对利尿剂抵抗的肺水肿。有趣的是,Hassoun等人将缺血再灌注(IR)-AKI模型的效果与双侧肾切除术进行了比较,发现IR会导致更严重的肺损伤。因此,AKI引起的肺损伤不太可能仅仅是由于肾功能下降引起,而受损肾脏释放的炎症介质可能是其原因之一。
在过去的几十年里,有越来越多的文献揭示了AKI过程中产生一些复杂的炎症介质,这些介质会导致肺损伤。病理生理学角度认为涉及细胞因子、损伤相关分子模式(DAMP)和其他促炎配体释放进入循环中,然后进入肺部并与肺组织上的受体结合。这些配体-受体的相互作用启动了促炎反应,导致血管通透性增加、免疫细胞渗透和非心源性肺水肿。中性粒细胞炎症是大多数类型肺损伤的特征,但在AKI期间,T细胞向肺的聚集也特别重要。已发现在IR-AKI后24小时T细胞在肺内聚集,而在有T细胞缺陷的小鼠中肺水肿减轻。
白介素6(IL-6)可能是AKI所致肺损伤最主要的介质,在临床前和临床研究中都有数据支持IL-6介导的炎症作用。例如Klein等人的研究结果表明,同样程度的肾功能受损,使用IL-6阻断抗体治疗的小鼠和IL-6基因敲除的野生型小鼠在IR-AKI后可免受肺损伤。IL-6水平的升高还与急性呼吸窘迫综合征(ARDS)和AKI患者的死亡率和机械通气持续时间增加有关。最后,研究已经证明,AKI后循环中的白介素8(IL-8)和肿瘤坏死因子(TNF)以及其他细胞因子水平会增加,并直接导致肺部炎症。
DAMPs是在应激状态下受损/死亡的细胞释放的内源性分子,研究认为可以从肾脏传递到肺部,在肺里发挥促炎作用。近期研究表明,线粒体DNA是IR-AKI后小鼠循环中增加的一种DAMPs,与新冠肺炎患者的肺损伤有关。还发现肾脏线粒体DAMPs直接导致了与肺部线粒体功能障碍一致的代谢变化。DeWolf等人研究表明,损伤的人原代肾小管上皮细胞产生的坏死液可以增加细胞因子/黏附分子的产生、丝裂原活化蛋白激酶(MAPKs)和核因子-κβ(NF-κβ)的激活以及增加血管通透性,导致体外培养的人微血管内皮细胞炎症增加。高迁移率蛋白族1(HMGB1)是另一种公认的DAMPs,也已被证明通过激活toll样受体4在AKI诱导的肺损伤啮齿类动物模型中发挥关键作用。
最后,Khamesi等人证明了骨桥蛋白是一种免疫调节分子,在AKI后在肾脏中产生,并进入到肺加重肺损伤。我们强调这篇优质的论文是因为它首次提供了明确的证据,证明肾脏是AKI后在肺中发挥作用的促炎分子的来源。其他细胞因子和DAMPs的细胞来源,包括上面提到的那些,还有待进一步证明。
当前管理呼吸机-肺-肾交互作用的方法
预防机械通气所致AKI的初始方法包括通过补液和用血管升压药来抵消正压通气对全身血液动力学的影响。然而,液体量和血管升压药的用量以及最适合肾脏保护的血管升压药尚未明确确定。此外,2006年的液体和导管治疗试验(FACTT)表明,采用保守的液体管理策略治疗的患者需要机械通气的天数更少,但是透析需求降低的趋势并不显著。就液体类型而言,有证据表明,平衡晶体液可降低对肾脏替代治疗的需求,也降低持续性肾功能不全的发生。需要注意的是,本研究中仅1/3的患者需要机械通气,与生理盐水相比,使用平衡晶体液不会影响无呼吸机天数。尽管如此,对于那些容量不足的患者,仍使用平衡晶体液进行液体复苏。尽管这缺乏数据支持可以改善肾脏的预后,但在实现充分液体复苏后,仍应遵循保守的液体管理策略。
去甲肾上腺素被认为是成人重症患者的一线血管加压药,但最近的研究显示,联合使用升压药如β-激动剂和血管紧张素II可能会改善肾脏预后。单独和联合使用特异性血管升压药的影响仍然是一个活跃的研究领域。前面提到由于机械通气导致的CVP增加对肾灌注产生有害影响,并且CVP增加与AKI发生有关。建议机械通气患者应设定更高的MAP目标以抵消高CVP的影响。然而,目前缺乏支持这一观点的数据,一般认为维持MAP大于65 mmHg就可以提供足够的肾灌注。
设置最佳的呼吸机参数来保护肺和肾是预防呼吸机引起的肺-肾损伤的另一个重要因素。目前的呼吸机管理方法是基于2000年的急性呼吸窘迫综合征网络(ARDSNet)试验,该试验表明,接受开放式肺保护性通气(低潮气量和高PEEP)治疗的ARDS患者的生存率和无肾衰竭天数增加。有趣的是,肺保护性通气对肾脏预后的改善程度超过了对心血管、肝脏、神经和血液系统的改善。虽然低潮气量通气明显具有肾保护作用,但关于最佳PEEP仍存在一些争议。前面的临床前和临床研究显示,高PEEP会导致肾功能障碍,但其他研究表明AKI风险与PEEP之间没有相关性。最近有研究表明,驱动压(平台压-PEEP)即肺顺应性,比单独的潮气量或PEEP更能预测患者的预后。然而,驱动压和AKI之间的关系尚未明确确定,最佳PEEP 与驱动压的关系仍有待确定。目前,大多数患者普遍采用基于理想体重的6–8 ml/kg的低潮气量通气,但最佳的肺-肾保护性呼吸机设置可能不是“一刀切”的。目前正在进行的研究重点是如何精准化以及患者个体化的设置呼吸机参数。
一旦发生AKI,治疗重点是减轻进一步损伤、避免使用肾毒性药物和使用适当剂量的血管活性药。2016年在ICU患者中进行的一项试验(包括80%接受机械通气的患者)中,早期肾脏替代治疗(RRT)并未改善死亡率。而且,延迟组中约一半的患者一直都不需要RRT。早期RRT可能会改善最终需要RRT患者的预后,但识别这些患者具有挑战性。最后,体外二氧化碳清除(ECCO2R)和体外膜肺氧合(ECMO)越来越多地用于支持危重患者的气体交换,但关于这些先进疗法在肺和肾保护方面的作用数据有限。
讨论
上述研究主要集中在机械通气对肾灌注和GFR的影响上,但这些功能变化如何转化为实质损伤的“结构性AKI”仍不清楚。这一概念很重要,因为如果氧利用率也降低,那么减少肾灌注或减少肾脏氧输送不一定会导致肾小管损伤和AKI。肾小管转运工作,特别是近端小管钠的重吸收,比任何其它肾功能都利用更多的氧气和ATP,肾小管转运工作与GFR直接相关。因此,在健康状态下,在低肾灌注的情况下,耗氧量会减少,从而保护肾脏免受缺氧和小管损伤。因此,在机械通气期间观察到的GFR降低可能具有保护作用。如果试图通过补液或血管加压药恢复GFR导致肾小管转运增加,也可能是对肾脏有害的。目前的研究仍缺乏对机械通气期间肾内生理变化与急性肾损伤之间关系,这些知识盲区也阻碍了新治疗方法的发展。
如上所述,机械通气与肾血流减少和钠重吸收增加有关。这些变化可能会引起小管转运功增加而减少氧输送并增加氧消耗,这可能会导致缺氧、代谢应激和细胞损伤。有趣的是,胰高血糖素样肽-1(GLP-1)激动剂已被证明可能通过抵消机械通气对肾损伤的方式影响肾脏生理。例如,已经发现GLP-1激动剂艾塞那肽除了在近端小管水平抑制钠的重吸收,还通过舒张肾内血管增加肾血流量来发挥肾保护的作用。支持这一观点的研究显示,在顺铂诱导的AKI的临床前模型中,GLP-1激动剂已被证明可以减少肾脏氧化应激和组织学损伤。在动物模型和I期临床试验中,还发现了其他针对肾脏代谢的治疗方法,如烟酰胺腺嘌呤二核苷酸(NAD+)增加,可预防AKI。这些治疗方法可能比目前通过补液或血管升压药保持肾血流量的方法更好,这方面的研究是有必要的。
最后,由于早期检测方面的挑战,预防AKI引起的肺损伤和肺损伤引起的AKI受到了限制。如前所述,如IL-6等炎症介质抑制剂,在临床前研究中显示出有希望的结果。IL-6抑制剂托珠单抗在治疗COVID-19中也显示出一些益处。但是大多数炎症介质在临床检测到AKI前就已经促进了肺和肾损伤。一些新的肾损伤生物标志物,如肾损伤分子-1(KIM-1)和胰岛素样生长因子结合蛋白7(IGFBP 7)已被证明可在血清肌酐升高前几小时预测中重度AKI,并且IGFBP 7的一系列测量已被证明对临床治疗有反应。这些生物标志物可能让一些可以让患者更早地进行抗炎治疗而获得益处。它们还可以用作提醒临床医生注意患者AKI和VILI风险增加的工具,可以更早地调整呼吸机模式或早期应用先进疗法(如ECCO2R或ECMO)。
结论
总之,机械通气期间的AKI亦或AKI期间的机械通气都是危重症的常见并发症,与较高的发病率和死亡率相关。除了低潮气量的肺保护性通气之外,目前几乎没有有证据支持的治疗。为了研究新型治疗方法,我们必须进一步了解机械通气期间肾脏生理改变与结构性AKI之间的关系。AKI影响呼吸力学和气体交换比容量超负荷作用大的机制也值得立即研究。最后,利用新型生物标志物预测机械通气和AKI相互需求的转化研究可能会促进更早的识别、临床特征和治疗。只有通过新颖的机制研究,才有可能发现新的治疗方法。

