发作性运动诱发性运动障碍(PKD):如何避免误诊?
时间:2024-09-23 15:01:19 热度:37.1℃ 作者:网络
论坛导读:发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia,PKD)是一种运动障碍,其特征为反复和短暂的不随意运动发作,包括肌张力障碍、舞蹈病、舞蹈症或这些症状的组合,通常由突然的随意运动触发。长期以来,基底神经节-丘脑-皮质回路的紊乱被认为是不自主运动的原因。基底神经节的门控功能受损会导致向丘脑的异常输出,这又会导致大脑皮层的过度激活。已经在PKD患者中发现基底神经节、丘脑和皮质的结构和功能异常以及这些脑区之间的异常连接。最近的研究强调了小脑在PKD中的作用。从小脑皮质到小脑深部核的抑制不足会导致丘脑皮质通路的过度兴奋。
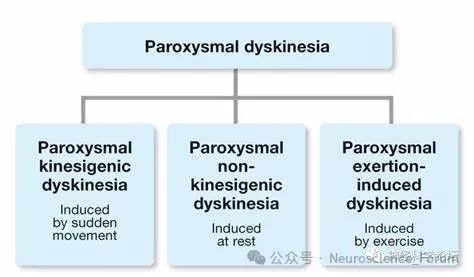
阵发性运动障碍是一种罕见的运动障碍,其定义为不自主的间歇性运动,可能包括肌张力障碍、舞蹈病或手足徐动症的任何组合。1885年,高尔出版了一本书,其中包括两个运动诱发癫痫的儿科病例的描述。随着时间的推移,由疲劳或运动引发的先兆发作的报告被发表,并且仅持续几分钟。发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia,PKD)是发作性运动障碍中最常见的类型,以突然运动诱发短暂的不自主运动为特征。由于相对少见,PKD易被误诊为癫痫或其他发作性疾病。
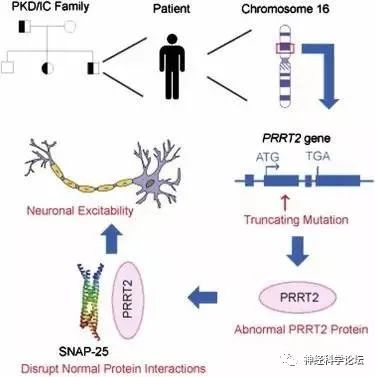
PKD 可分为家族性和散发性,其中家族性PKD呈常染色体显性遗传(B级证据),国内学者利用全外显子测序结合Sanger测序,在国际上首次发现PRRT2基因(NM_145239.2)是其致病基因,之后国内外学者陆续证实了这一结论(B 级证据)。PRRT2 基因突变多为移码突变,其中 c.649dupC 为热点突变,存在外显不全 (incomplete penetrance)及 新 发 突 变(de novo)现 象(B 级证据)。近期,国内学者又报道了 PKD 的第二个致病基因TMEM151A。由于 PKD 相对少见,临床上容易误诊为癫痫或其他疾病。2004 年,PKD 的临床诊断标准就 已提出,但关于PKD 治疗的循证医学证据极少。2013年,国内一项横断面临床队列研究结果提示,口服小剂量卡马西平可以完全控制携带 PRRT2基因突变患者的运动障碍,为PKD的治疗提供了临床证据并得到进 一 步证实(B级证据)。
PKD又称发作性运动诱发性舞蹈手足徐动症(paroxysmal kinesigenic choreoathetosis),在1967年被首次报道并命名,以 静止状态下突然运动并诱发出短暂的不自主运动 为特征,包括舞蹈症、肌张力障碍、手足徐动症、投掷症等。PKD 是发作性运动障碍中最常见的类型,多在儿童期发病,青春期时发作频率最高,严重影响青少年的身心健康。
Neurosci Bull. 2024 Jul;40(7)
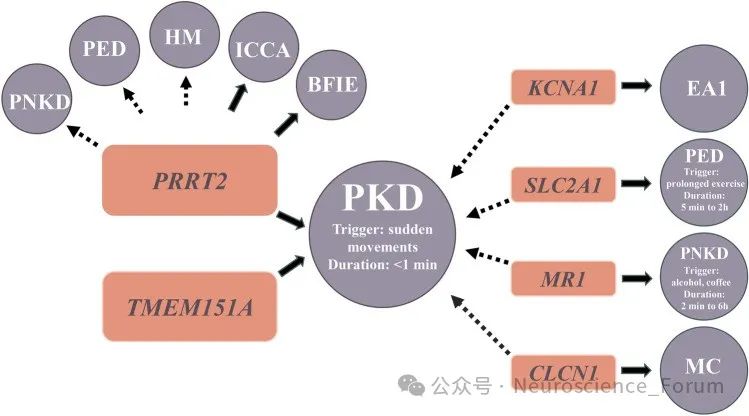
PKD分为原发性和继发性,原发性PKD主要是由于PRRT2和TMEM151A基因突变导致,极少数患者亦可能检出KCNA1基因突变。继发性PKD原因包括多发性硬化、头部外伤、假性甲状旁腺功能减退(PHP)等, iPKD的发病机制口前尚不完全清楚。研究结果显示PRRT2是一种新的突触蛋白,可影响神经元突触内的SNARE蛋白复合体形成。PRRT2基因突变影响了小脑内颗粒细胞的平行纤维与浦肯野细胞之间的突触传递,造成浦肯野细胞异常放电,进而导致小脑功能紊乱和发作性运动障碍。近期有研究结果提示PRRT2基因突变会导致小脑颗粒细胞钠离子通道活性增强和小脑皮质对去极化扩布(spreading depolarization)易感,提出了发作性运动障碍的“小脑去极化扩布”假说,为去极化扩布的敏感性调控提供了新的分子机制。
与突触功能有关的PRRT2(富含脯氨酸的跨膜蛋白2)基因突变已被确定为一系列疾病的原因,包括:阵发性运动性运动障碍(PKD)、良性家族性婴儿癫痫、发作性共济失调、偏瘫性偏头痛和复杂的神经发育障碍。PRRT2基因突变,占绝大多数孤立性PKD病例(27%至65%的病例)。这种表现型以短时间(少于1分钟)发作为特征,包括由突然运动、移动意图和/或加速触发的舞蹈病、张力障碍和/或弹道运动。并发的抽动障碍在PKD患者中常常会被忽略,而抽搐与PKD有着共同的未知病理生理机制。研究已经提出抽动障碍、PKD和癫痫之间可能存在共同的机制,最近的一项研究表明阵发性运动障碍之间可能存在共同的遗传易感性。
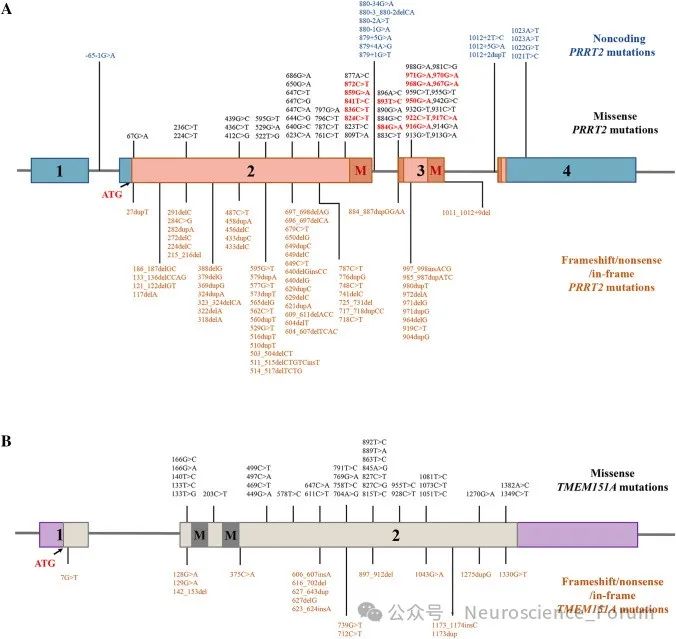
PRRT2错义突变的功能研究和致病性分类表明,所有可能的致病性突变都聚集在PRRT2的C-末端。PRRT2基因编码340个氨基酸的跨膜蛋白,在大的细胞间N-末端区域具有富含脯氨酸的结构域,在C-末端区域具有两个推定的跨膜结构域。致病性错义突变导致PRRT2从质膜到细胞质的亚细胞错定位。这种错位可能与位于C末端的受损跨膜结构域有关。此外,没有完整C-末端的截短突变导致与蛋白酶体介导的截短蛋白降解相关的PRRT2蛋白水平降低。PRRT2有缺陷的C末端与PKD密切相关。
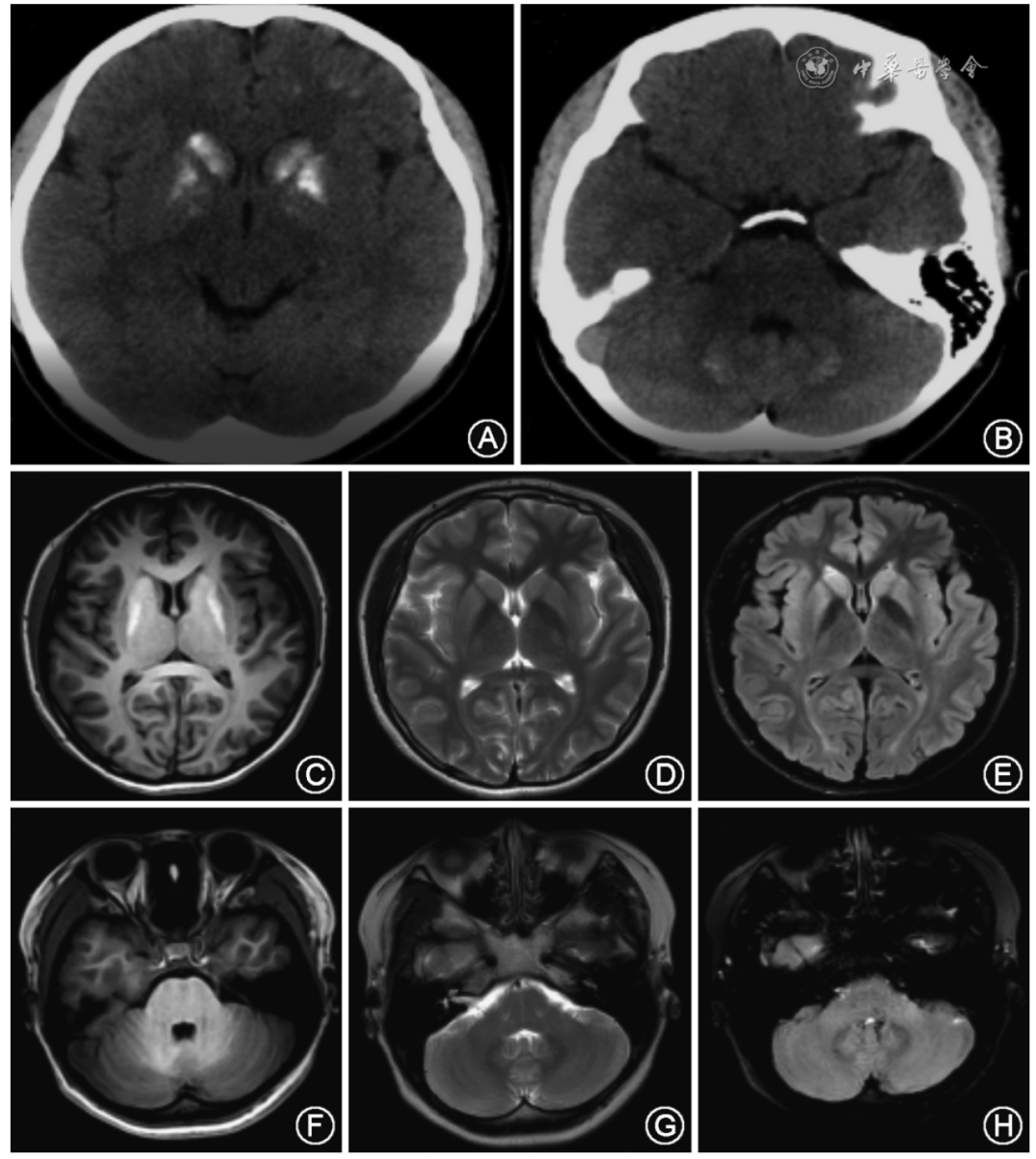
中华神经科杂志, 2023,56(10)
PHP是继发性PKD的罕见原因,目前国内外仅有9篇文献报道,具体的病理生理机制尚不清楚。有研究者认为,由于离子通道紊乱促使神经肌肉过度兴奋导致PKD发作性症状。细胞外钙离子水平下降,可使阈电位下降,向静息电位水平靠近,细胞的兴奋性升高,故PHP导致的低钙血症容易促使神经肌肉兴奋性增高。卡马西平、奥卡西平等钠通道阻滞剂药物可降低细胞膜对钠离子和钙离子的通透性,从而降低细胞的兴奋性。临床上使用卡马西平、奥卡西平等钠通道阻滞剂药物治疗PKD可取得良好的疗效,这也支持PKD可能是一种离子通道病的观点。PHP导致的低钙血症可以影响含有γ-氨基丁酸(γ-aminobutyric acid,GABA)的突触小泡的活性,使其对中枢神经元抑制作用减弱,而促使其他中枢神经元兴奋性相对增加,神经肌肉兴奋性增高,从而出现运动障碍。
Neurosci Bull. 2024 Jul;40(7)
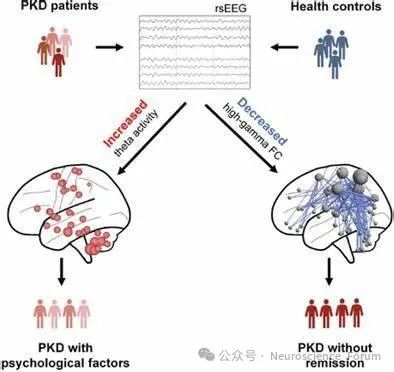
PKD的发病机制很复杂,目前对它的了解还不完全。PRRT2基因被鉴定为PKD的主要致病基因。神经元中的离子通道、离子转运蛋白和突触传递都受到缺陷型PRRT2的影响。其中,与Na+通道的相互作用很好地解释了为什么PKD患者对CBZ有极好的反应,并有力地证实了PKD是一种离子通道病。突触传递中的启动和神经递质释放过程也受到PRRT2的影响,但迄今为止不同研究的实验结果并不一致。突触疾病的猜测需要通过处理相互矛盾的实验室结果来验证。值得注意的是,近年来小脑被认为是产生运动障碍的主要部位。然而,在PKD的进展中,异常的基底神经节-丘脑皮层回路和小脑-丘脑回路经常被神经影像学方法发现。考虑到通过神经影像学方法也可以在PKD患者中发现继发性改变或代偿性改变,而不是根本原因,需要在适当的生物PKD模型中进一步确定PKD发病机制中的这些脑区。
2020年,我国PKD诊治领域的专家经讨论形成了 PKD 诊治英文版专家共识。

修订后的原发性 PKD 临床诊断标准

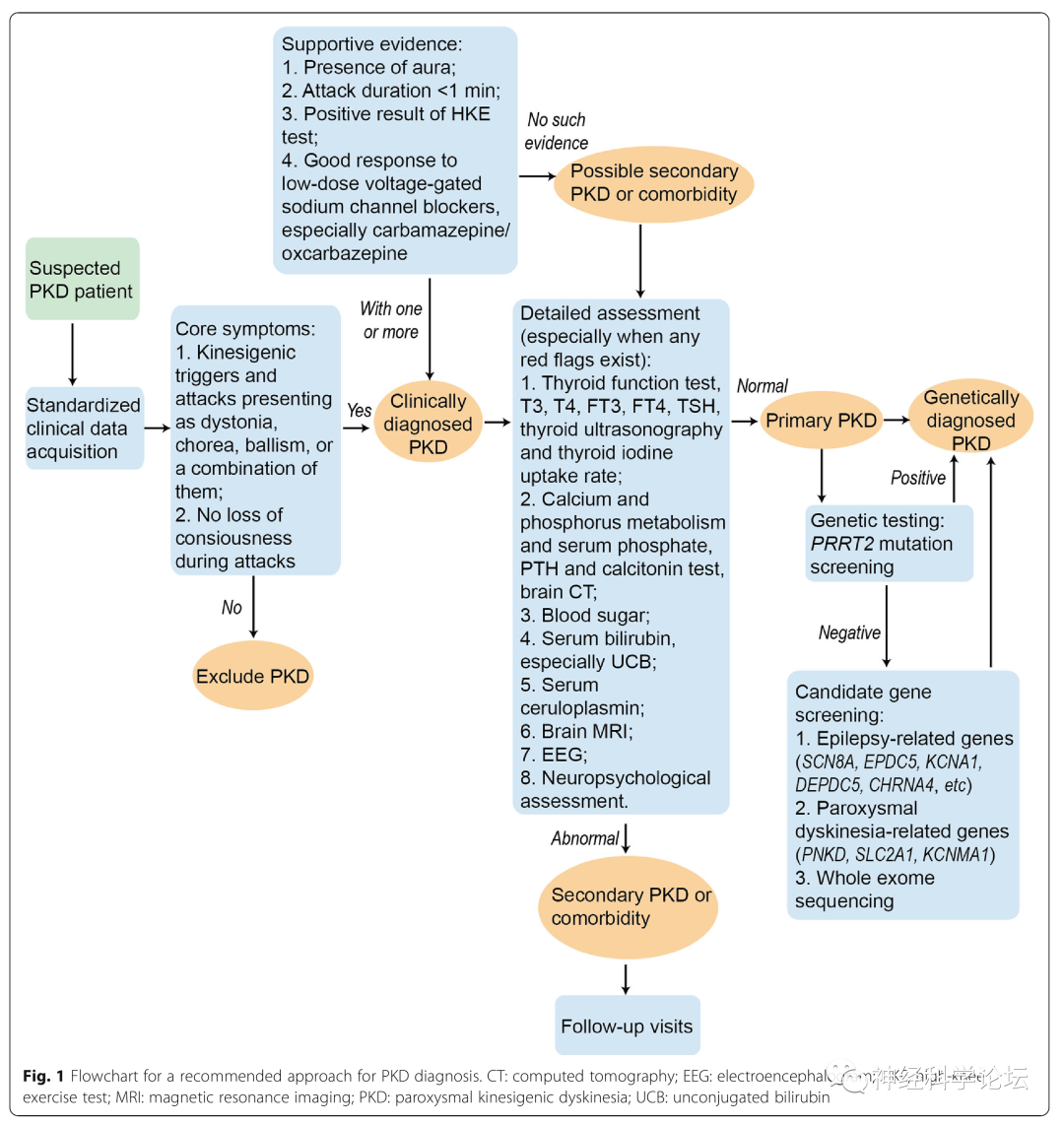
PKD诊断推荐方法的流程图
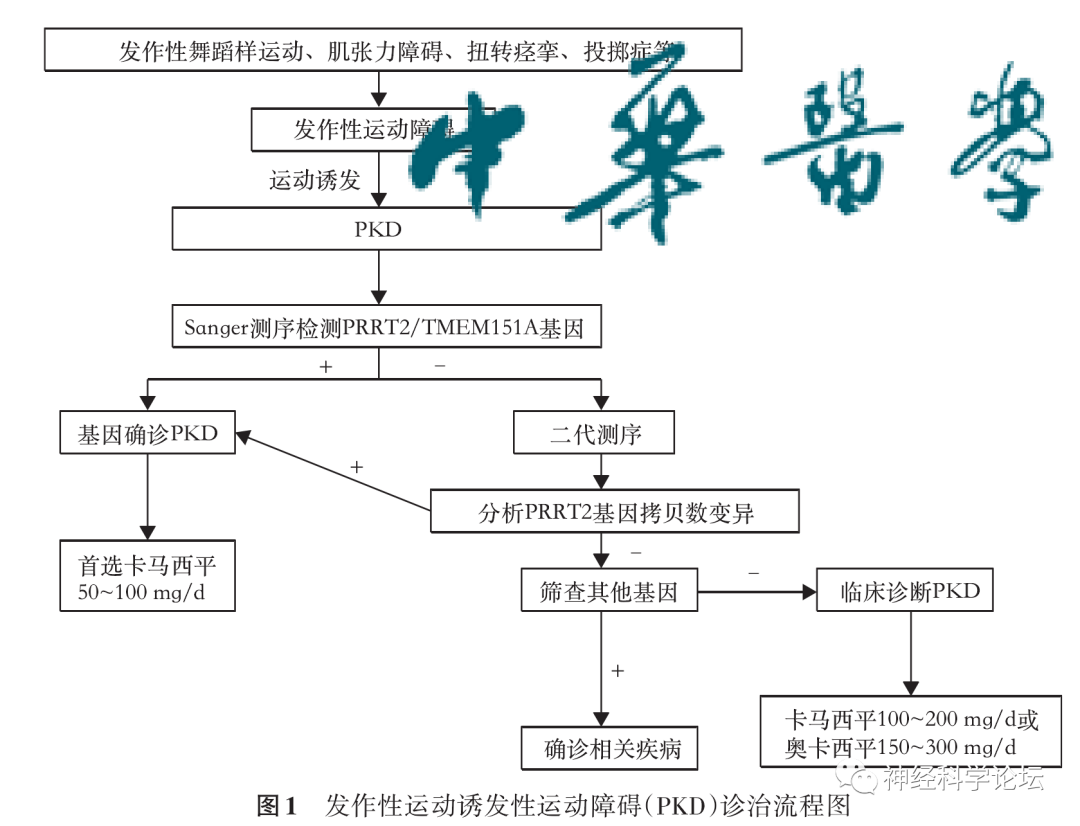
为进一步提高各级临床医生对 PKD 的认识和诊治水平,制定了中文版指南并归纳出 PKD 的诊治流程图并发表在中华神经科杂志上。

PKD的诊断标准包括已确定的发作运动诱发因素、持续时间短(< 1分钟)、发作期间无意识丧失或疼痛、神经学检查正常并排除了其他鉴别诊断、抗癫痫治疗(如卡马西平和苯妥英)后临床症状改善以及发病年龄在20岁之前。10%的PKD儿童可能出现这些事件之前的先兆。6或者,可以证明PRRT2基因的杂合显性突变支持诊断。该儿童报告的致病性遗传变异——prr T2 c . 649 dup p .(arg 217 fs)导致翻译过早终止。此外,文献中还报道了它与PKD的关系。虽然在几年前首次被描述,但普通儿科医生和其他临床医生仍然缺乏认识。一项研究显示,获得正确诊断的平均时间为4.8年。患者通常很难清楚地识别触发因素,相关的焦虑可能被报告为一个原因。这可能导致误诊,如心身障碍,甚至抽搐障碍。
-
对疑似抽搐样或舞蹈病的病史采集应包括探索“突然运动”是否是可能的触发因素。
-
当临床医生对复杂或不典型的诊断持有不确定性时,频繁复查、重复临床检查和延长视频片段是有价值的。
-
临床医生应该寻求理解情绪困扰与诊断的联系。焦虑可能是一种状况的结果,而不是原因或相关症状。
询问关于突然运动可能是“假定的抽搐样障碍”的触发因素的引导性问题是有用的。鉴于苯妥英副作用较小,卡马西平是治疗PKD的首选药物。大多数儿童对治疗反应积极,随着年龄的增长,事件发生的频率降低。PKD是一个具有挑战性的诊断,认识到它是一个运动诱发的肌张力障碍是关键。在这种情况下,识别情绪困扰以及它与诊断的关系也很重要。此外,如果临床医生保持开放的心态,反复的临床回顾要么给诊断带来确定性,要么突出缺陷区域。这允许随着额外信息的获得而发展诊断,并最终为患者和家属提供正确的结果。
参考文献
Li ZY, Tian WT, Huang XJ, Cao L. The Pathogenesis of Paroxysmal Kinesigenic Dyskinesia: Current Concepts. Mov Disord. 2023 Apr;38(4):537-544. doi: 10.1002/mds.29326.
Yoo D, Kim HJ, Choi JH, Lim JH, Jeon B. Tics in Paroxysmal Kinesigenic Dyskinesia. Mov Disord Clin Pract. 2019 Jun 6;6(6):502-503. doi: 10.1002/mdc3.12779.
Recommendations for the diagnosis and treatment of paroxysmal kinesigenic dyskinesia: an expert consensus in China.Transl Neurodegener . 2021 Feb 16;10(1):7.
Cottrill N, McCully B, Payne M. Paroxysmal Kinesigenic Dyskinesia Presented Following Concussion. J Mov Disord. 2019 Jan;12(1):52-53. doi: 10.14802/jmd.18027.
Xu JJ, Li HF, Wu ZY. Paroxysmal Kinesigenic Dyskinesia: Genetics and Pathophysiological Mechanisms. Neurosci Bull. 2024 Jul;40(7):952-962. doi: 10.1007/s12264-023-01157-z.
中华医学会神经病学分会神经遗传学组等.中国发作性运动诱发性运动障碍诊治指南.中华神经科杂志,2022,55(1):9-14.
陈秋蕾,翁诗雯,等.假性甲状旁腺功能减退症导致发作性运动诱发性运动障碍2例并文献复习.中华神经科杂志, 2023,56(10) : 1119-1127.
Sanpera J, Gupta R, Singh R, Byrne S. PRRT2- Associated Paroxysmal Kinesigenic Dyskinesia Only Evident with High-Speed Cricket Bowling. Mov Disord Clin Pract. 2022 Jan 8;9(2):259-260. doi: 10.1002/mdc3.13402.
