J Clin Invest 浙大二院王建安院士/胡新央教授团队揭示FMO2改善缺血后心肌损伤新机制
时间:2024-11-07 06:00:13 热度:37.1℃ 作者:网络
心肌梗死(MI)是全球主要的致死原因之一。尽管再灌注策略和药物治疗取得了重要进展,MI仍对全球健康构成严重威胁。在缺血期间,大量心肌细胞死亡被认为是导致心肌损伤的重要病理过程,并进一步促使心力衰竭的发生。然而,MI后心肌细胞死亡的基本机制尚未完全阐明,依然缺乏有效的靶向治疗手段。
2024年10月31日,浙江大学医学院附属第二医院王建安院士/胡新央教授团队在Journal of Clinical Investigation杂志上发表了题为“Flavin-containing monooxygenase 2 confers cardioprotection in ischemia models through its disulfide-bond catalytic activity”的研究论文。该研究首次揭示了含黄素单加氧酶2 (FMO2)作为心肌细胞中参与蛋白质氧化折叠的关键调节因子,为缺血性心脏病的治疗提供了新的策略和干预靶点。

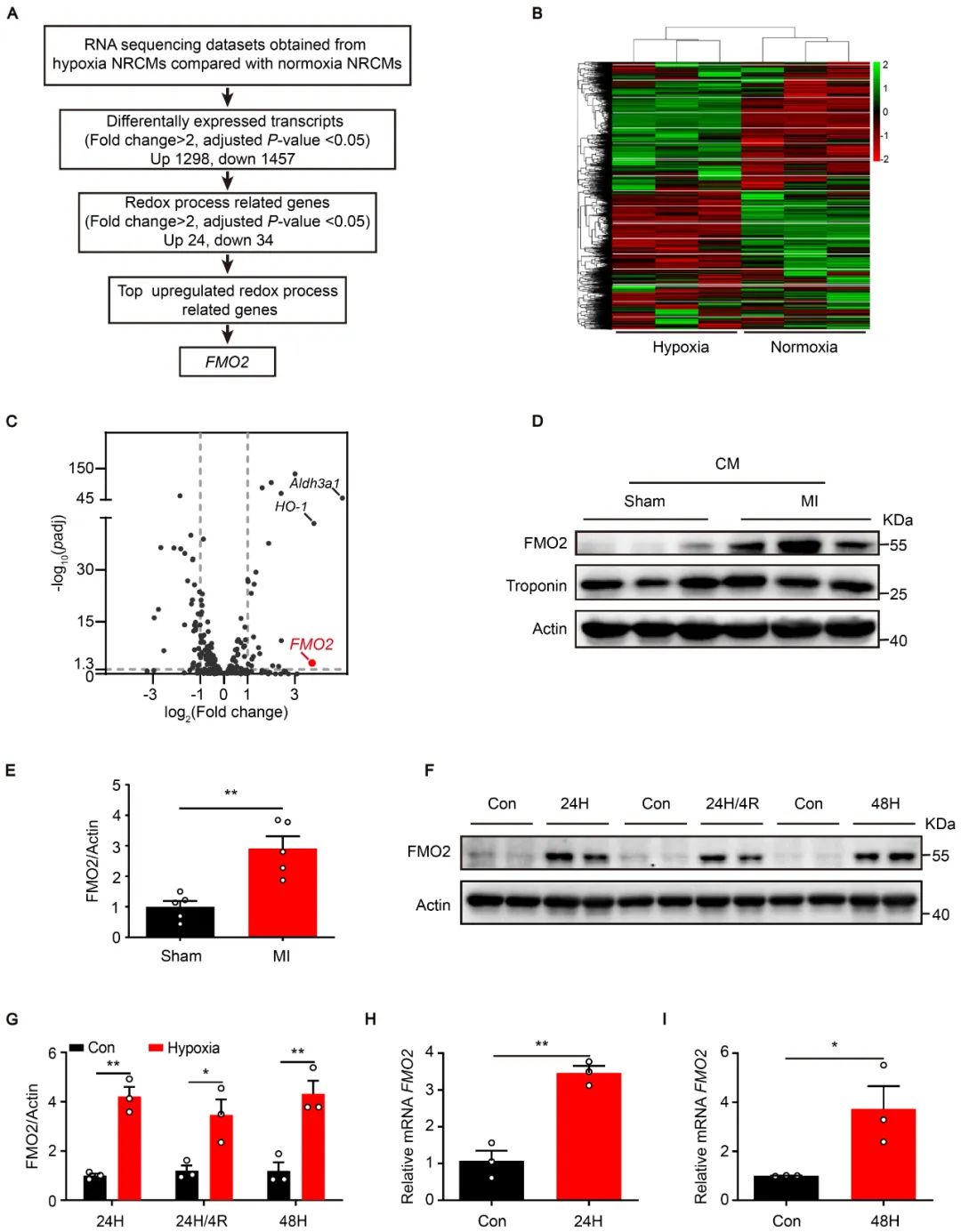
首先,作者对常氧和缺氧条件下的新生大鼠心肌细胞(NRCMs)进行高通量转录组测序分析,发现参与氧化还原过程的基因中,FMO2升高最为显著。在分离的大鼠心梗后心肌细胞中,FMO2蛋白表达明显增加。同时,在培养的NRCMs中,缺氧处理后FMO2的蛋白和mRNA水平显著上调。
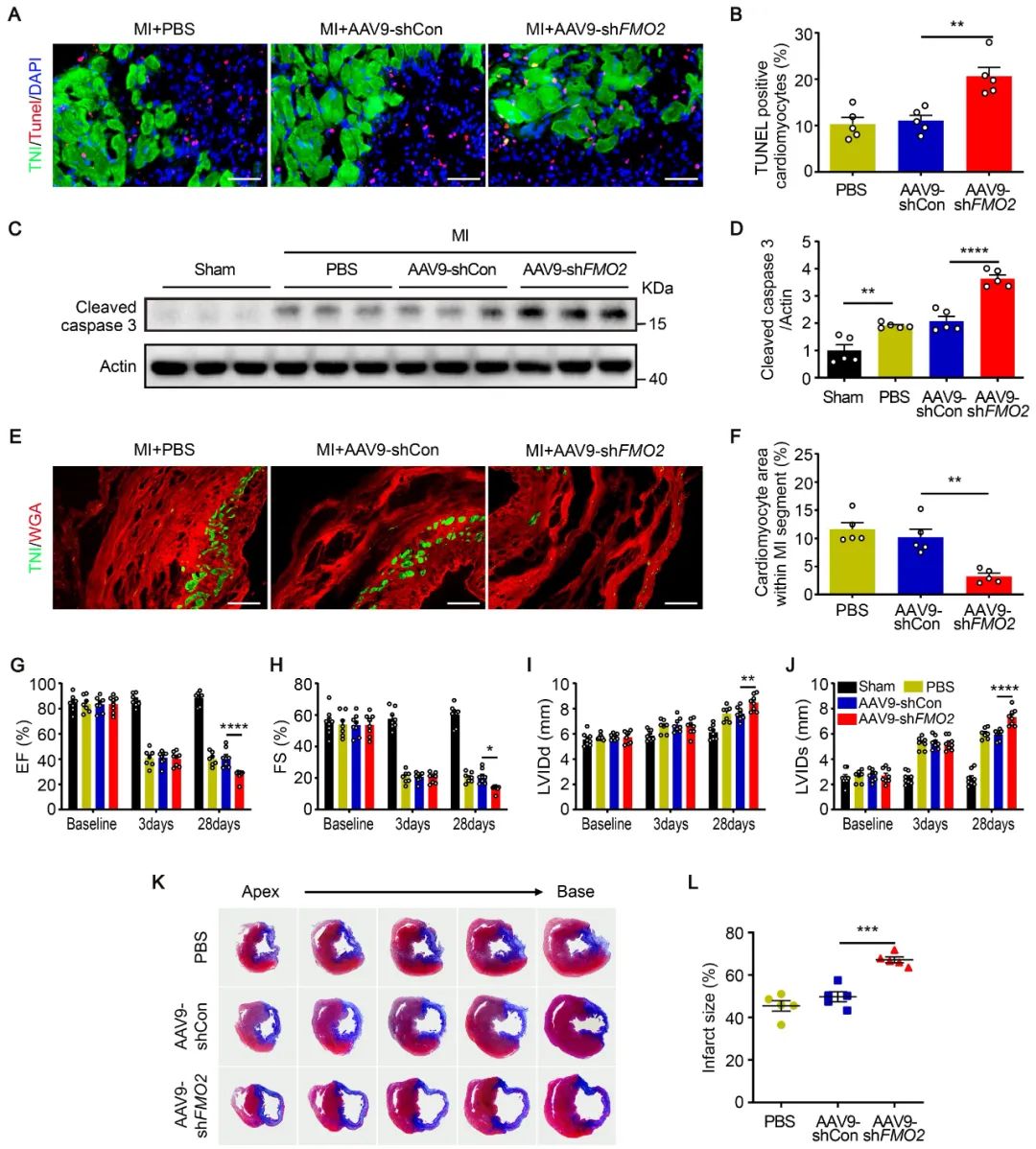
为研究FMO2在心肌缺血损伤中的作用,作者利用腺相关病毒载体构建心肌细胞特异性敲减或表达FMO2的大鼠模型。在MI术后第3天,FMO2敲减组心脏梗死周边区心肌细胞凋亡增加;在MI后第28天,敲减组的心脏存活心肌细胞数量减少,心功能下降且瘢痕面积增大。而心肌细胞特异性过表达FMO2则表现出显著的心脏保护作用。
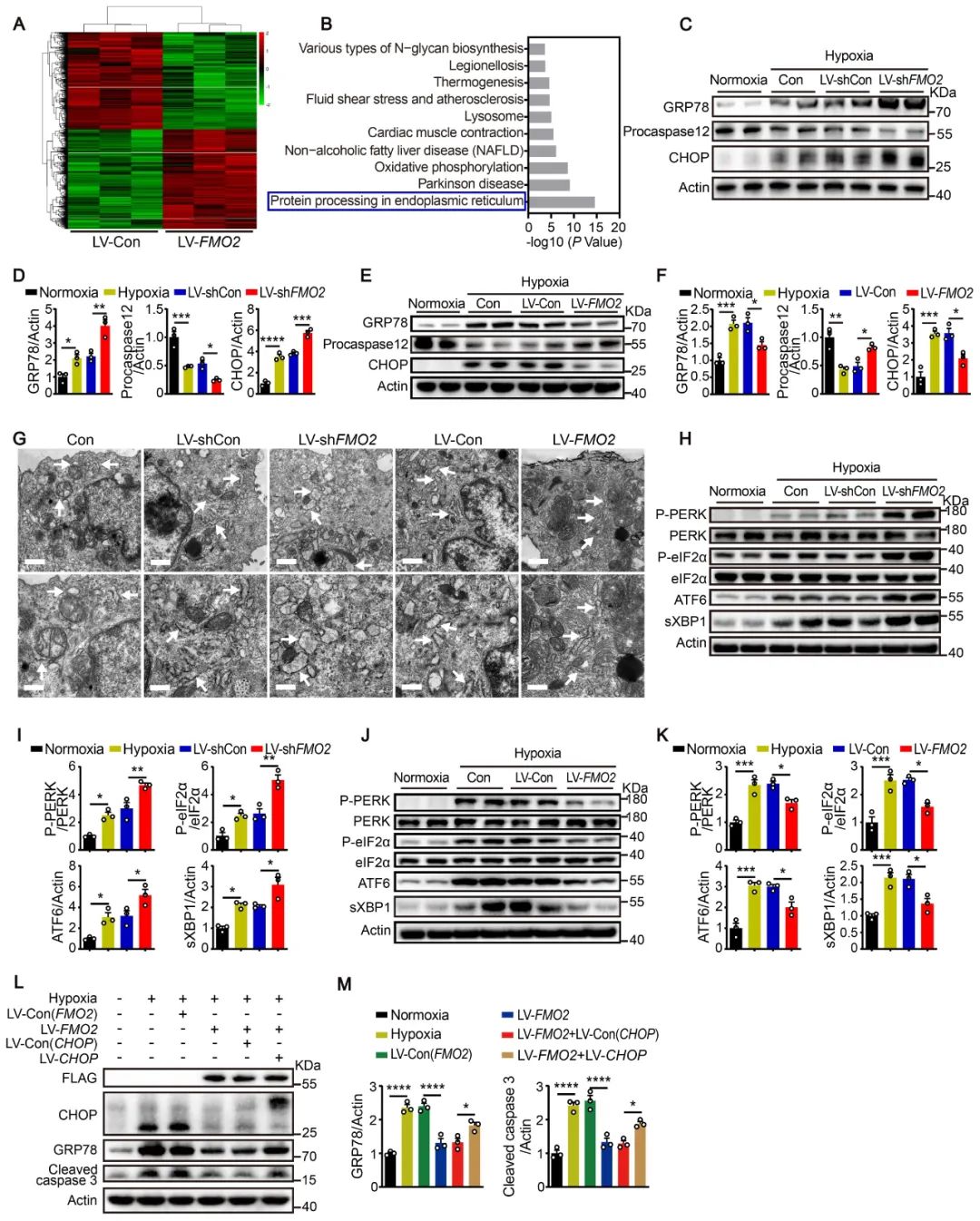
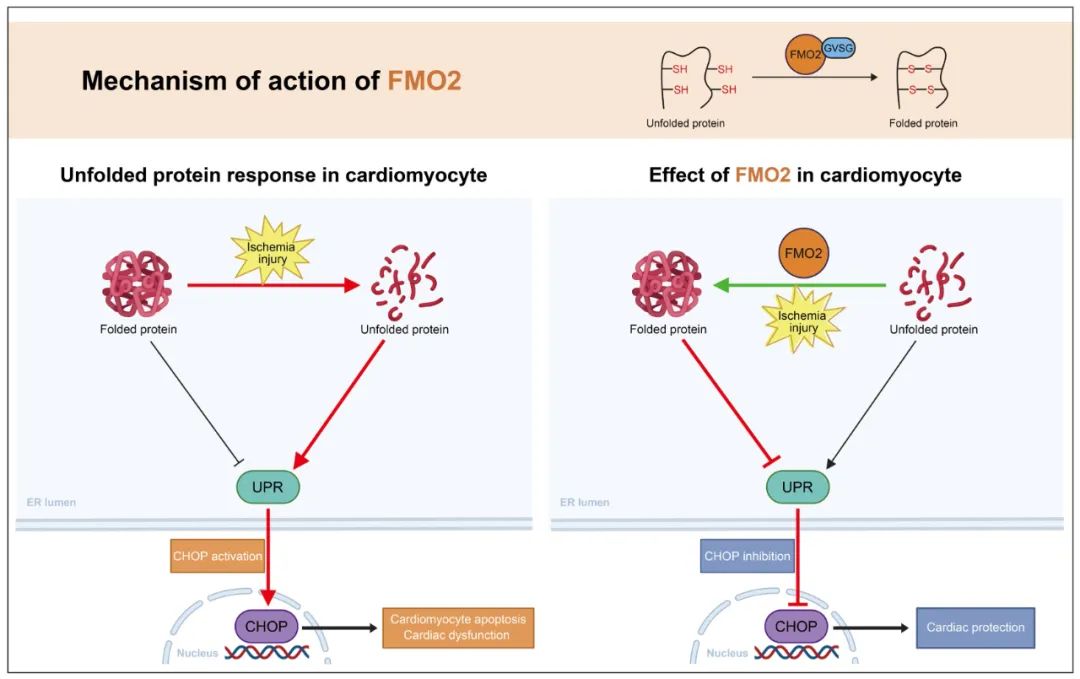
为了探究FMO2介导的心肌保护机制,作者对过表达FMO2的心肌细胞进行高通量转录组测序分析。KEGG富集分析显示,“the protein processing in endoplasmic reticulum pathway”最为相关。研究发现,敲减FMO2会加重内质网应激,表现为78kDa葡萄糖调节蛋白(GRP78)、C/EBP同源蛋白(CHOP)和cleaved caspase 12的蛋白表达上调,而过表达FMO2则产生相反的效果。此外,敲减FMO2会上调未折叠蛋白反应(UPR)通路蛋白激酶RNA样内质网激酶(PERK)、剪切型X-box结合蛋白1 (sXBP1)和激活转录因子6 (ATF6)的表达,而过表达FMO2抑制UPR。这些结果表明,FMO2可能通过UPR通路减少心肌细胞凋亡。
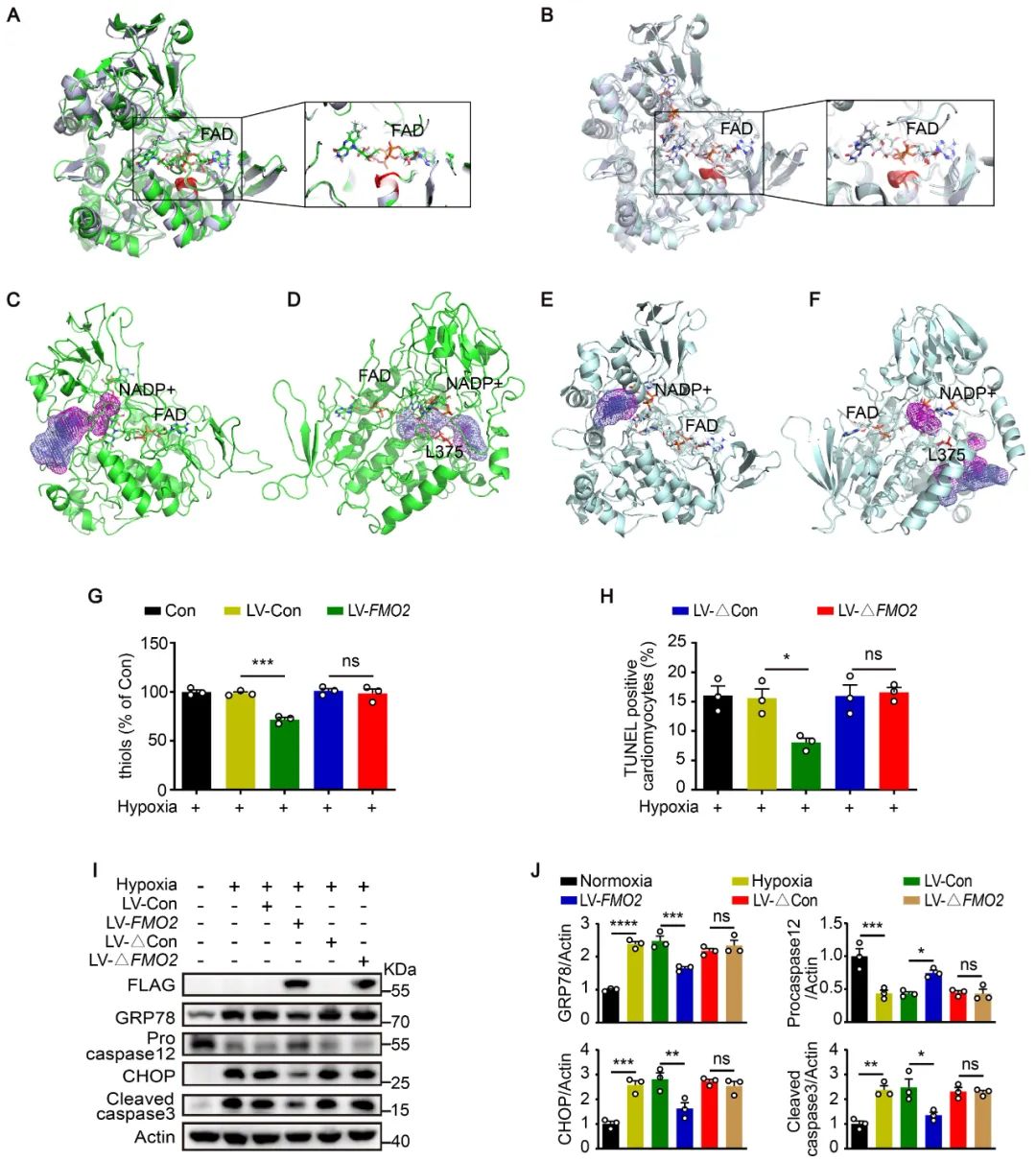
FMO2的表达显著增强了心肌细胞对二硫键还原剂DTT诱导的细胞毒性的抵抗力。利用5,5'-二硫代双(2-硝基苯甲酸)(DTNB),作者发现过表达FMO2的NRCMs中巯基水平较低,而敲减FMO2的细胞中巯基水平更高。FMO2蛋白可以直接催化还原性谷胱甘肽(GSH)内的巯基形成二硫键(氧化性谷胱甘肽GSSG),并且可以催化十肽NRCSQGSCWN上的半胱氨酸残基形成二硫键。这些结果表明,FMO2能够催化二硫键的形成。通过结构分析等方法,作者发现FMO2依赖其GVSG基序催化未折叠/错误折叠蛋白中的二硫键形成。体内外实验结果均表明,GVSG突变的FMO2无法催化二硫键形成,失去对内质网应激和UPR介导的心肌细胞凋亡的保护作用。
综上所述,此项研究表明FMO2依赖其酶活性,通过GVSG基序催化二硫键形成,从而抑制UPR途径,减轻心梗后心肌细胞死亡和心功能障碍,揭示了FMO2在治疗缺血性心脏病中的重要治疗潜力。
浙江大学医学院附属第二医院心血管内科医师刘庆年、黄继钮、丁豪、干部保健科陶悦为本文共同第一作者,王建安院士、胡新央教授为共同通讯作者。
原文链接:
https://www.jci.org/articles/view/177077

