Nat Commun:南京医科大学沈洪兵/马红霞/王铖合作研究为推进NSCLC风险分层提供了策略
时间:2025-02-27 12:11:32 热度:37.1℃ 作者:网络
全基因组关联研究已经确定了数千个与非小细胞肺癌(NSCLC)相关的遗传变异,然而,确定致病变异和改进疾病风险预测仍然具有挑战性。
2025年2月6日,南京医科大学沈洪兵、马红霞及王铖共同通讯在Nature communications 上在线发表题为“Massively parallel variant-to-function mapping determines functional regulatory variants of non-small cell lung cancer”的研究论文。研究应用大规模平行报告基因检测来进行NSCLC变异体到功能的大规模定位。
共评估了1249个候选变异,并在12个基因座中确定了30个潜在的因果变异。因此,研究提出了NSCLC易感性的三种遗传结构:单个单倍型块中的多个因果变异(例如4q22.1)、多个单倍型块中的多个因果变异(例如5p15.33)和单个因果变异(例如20q11.23)。研究使用来自中国人群的潜在因果变异开发了改进的多基因风险评分,提高了来自英国生物样本库的450,821名欧洲人的风险预测性能。研究结果不仅增强了对NSCLC易感性遗传结构的理解,而且还为推进NSCLC风险分层提供了策略。

肺癌在中国和世界范围内均具有较高的发病率和死亡率。非小细胞肺癌(NSCLC)占肺癌病例总数的85%,对公共卫生构成重大威胁。NSCLC是由环境暴露和遗传的生殖细胞系遗传变异体驱动的多因素疾病。全基因组关联研究(GWAS)是识别NSCLC遗传因素的有效方法。迄今为止,大规模GWAS已经在不同人群中鉴定了数千个具有全基因组意义的NSCLC遗传变异。然而,确定在GWAS中表现出表型的因果变异仍然很困难。超过90%的已鉴定遗传变异位于基因组的非编码区域,功能未知。此外,由于连锁不平衡(LD),这些位点中的每一个都可以包含数百个与疾病相关的单核苷酸多态性(SNP),这使得确定那些在功能上有助于表型的遗传变异变得极其困难。
已经开发了多种精细定位策略来确定这些致病变异。一种流行的策略是通过将包含LD信息的统计关联与表观遗传注释相结合来提出遗传机制的假设。然后,通过低通量实验提名少量候选变体进行验证。通过采用该策略,最近的研究成功地确定了多种疾病的一系列因果变异,但他们中的大多数一次研究一个基因座,由于功能评估变异的数量有限,几乎无法阐明所有因果变异或阐明每个基因座的遗传结构。大规模平行报告基因检测(MPRA)能够对非编码DNA序列的转录调控潜力进行高通量实验评估。在MPRA中,将多个调节元件克隆到包含报告基因和独特DNA条形码的表达载体中,从而创建表达文库。然后使用高通量测序分析该文库,以评估克隆元件的调节活性。迄今为止,MPRA已成功识别多种性状和疾病的致病变异,但它们尚未被系统地用于肺癌研究。
在这项工作中,设计并应用了MPRA来系统表征之前研究中NSCLC GWAS基因座下的致病变异,其中包括中国人群中的14,240例病例和14,813例对照个体。总共评估了1249个遗传变异,并在3种肺相关细胞类型中鉴定了82个功能调节变异(frVars)。通过整合MPRA和肺特异性转录调节注释,研究在12个位点中确定了30个潜在的因果变异,揭示了NSCLC易感性的不同遗传结构。研究还使用肺组织特异性表达数量性状基因座(eQTL)数据库确定了这些变异的靶基因,并阐明了三个代表性基因座(4q22.1、5p15.33和20q11.23)的遗传机制。最后,研究将潜在的因果变异纳入多基因风险评分(PRS)的构建中,并在来自英国生物样本库(UKB)数据集的450,821名欧洲人的队列中对其进行评估,旨在提高多基因风险预测的交叉祖先性能。
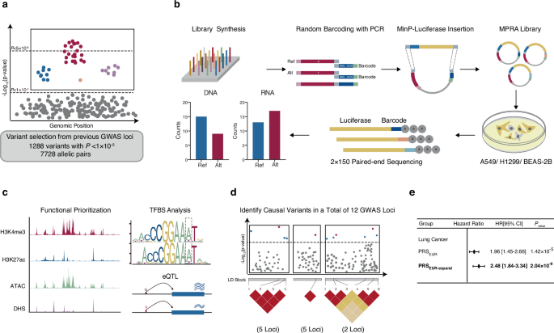
图1 研究概述(摘自Nature communications )
参考消息:
https://doi.org/10.1038/s41467-025-56725-w

