Cell Stem Cell:陈建军/苏瑞/沈柏用/田景琰团队发现TET2调控白血病干细胞归巢以及干性维持的新机制
时间:2023-08-07 19:30:59 热度:37.1℃ 作者:网络
TET2在急性髓性白血病(AML)中反复突变,其缺乏促进白血病发生(由侵袭性致癌突变驱动)并增强白血病干细胞(LSC)的自我更新。然而,潜在的细胞/分子机制尚未完全了解。
2023年8月3日,美国希望之城国家医疗中心陈建军、苏瑞及上海交通大学的沈柏用、田景琰共同通讯在Cell Stem Cell 在线发表题为“TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal”的研究论文,该研究发现Tet2缺乏通过促进LSCs归巢到骨髓(BM)生态位以增加其自我更新/增殖,显著促进各种AML模型(由侵袭性或非侵袭性突变介导)的白血病发生。
AML母细胞中TET2缺失增加了Tetraspanin 13 (TSPAN13)的表达,从而激活了CXCR4/CXCL12信号,导致LSCs增加了向BM生态位的归巢/迁移。在机制上,TET2缺乏导致TSPAN13 mRNA中甲基-5-胞嘧啶(m5C)修饰的积累;YBX1特异性识别m5C修饰,增加TSPAN13转录本的稳定性和表达。总的来说,该研究揭示了TET2作为mRNA m5C去甲基化酶在白血病发生、白血病母细胞迁移/归巢和LSC自我更新中的功能重要性。

在正常的造血系统中,基因表达不仅受到基因增强子和启动子的严格调控,还受到表观遗传和表转录组修饰的严格调控。越来越多的证据表明,DNA或RNA共价化学修饰的失调导致造血恶性肿瘤,包括急性髓性白血病(AML)。例如,在15%-20%的AML、30%的骨髓增生异常综合征(MDS)和50%的慢性髓单核细胞白血病(CMML)病例中发现TET甲基胞嘧啶双加氧酶2 (TET2)的体细胞耗尽和功能缺失突变。TET2催化5-甲基胞嘧啶(5mC)转化为5-羟基/甲酰基/羧基胞嘧啶(5hmC、5fC和5caC)介导DNA去甲基化,在转录水平上调控基因表达起核心作用。
绝大多数TET2突变失去去甲基化活性,被认为是AML髓系恶性转化的第一步。此外,TET2缺乏导致AML患者预后不良和耐药。12-16除了DNA 5mC修饰的氧化,最近的研究支持TET2介导的RNA(包括信使RNA (mRNA))中甲基-5-胞嘧啶(称为“m5C”以区别于DNA 5mC)的去甲基化的新功能。然而,对mRNA m5C的作用和生物学相关性的系统研究仍处于起步阶段。目前还完全不清楚TET2诱导的mRNA m5C去甲基化是否以及(如果有的话)如何促进白血病的发生或肿瘤的发生。
AML是最常见和最具侵袭性的血液恶性肿瘤之一,其特点是未成熟白血病干细胞(LSCs)的克隆扩增。尽管目前有可用的治疗方法,但超过70%的AML患者存活时间不超过5年。复发仍然是治疗失败的主要原因,超过50%的AML患者复发LSCs处于AML细胞层级的顶端,被认为是耐药和AML复发的根本原因。因此,根除LSCs对于治疗AML至关重要。LSCs存在于骨髓(BM)中称为“壁龛”的特殊微环境中,以支持其生存并维持其自我更新能力众所周知,趋化因子受体4 (CXCR4)与其配体CXCL12(也称为基质细胞衍生因子-1,SDF-1)之间的直接相互作用引发了微环境中LSCs和BM基质细胞之间的串扰。药理抑制CXCR4可阻止AML细胞的锚定,并促进LSCs动员出内皮生态位,从而改善其对化疗药物的易感性。
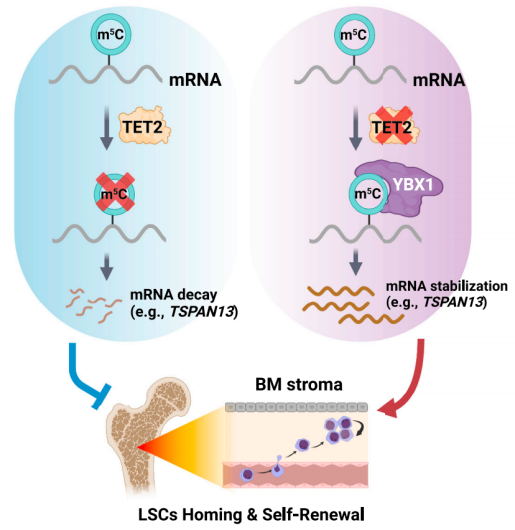
机理模式图(图源自Cell Stem Cell)
TET2在造血分化和LSC维持中的功能已经得到了很好的证明,但其潜在的细胞/分子机制仍然难以捉摸。尽管先前的研究表明TET2缺乏促进了侵袭性突变驱动的白血病发生,如混合谱系白血病(MLL)-AF9和AML1-ETO9a,但TET2缺乏对其他侵袭性较低突变驱动的AML模型的影响尚未确定。此外,TET2在BM微环境/生态位规划中的作用尚未被研究。
该研究发现TET2作为RNA去甲基化酶,介导四跨蛋白13 (Tspan13) mRNA稳定性的转录后调控。因此,Tet2缺乏导致Tspan13/Cxcr4轴的激活,进而导致LSC归巢/自我更新增加,从而促进白血病的发生。总的来说,该研究揭示了TET2在AML发病机制中作为RNA去甲基化酶调节BM生态位编程和LSC归巢/自我更新的作用和潜在的分子机制。
美国希望之城国家医疗中心陈建军教授、苏瑞助理教授以及上海交通大学沈柏用教授、田景琰教授为这一研究成果的共同通讯作者。希望之城国家医疗中心的黎扬婵博士、薛美琳博士、邓晓岚博士和董磊博士为本文的共同第一作者。
原文链接:
https://www.cell.com/cell-stem-cell/fulltext/S1934-5909(23)00246-1

