Nat Cardiovasc Res 张玉霞/顾晓琼/吴玉章合作揭示NO信号通过MCM8介导的线粒体自噬维持心血管功能的新机制
时间:2023-08-15 19:34:29 热度:37.1℃ 作者:网络
川崎病(KD)是一种全身性的血管炎,是5岁以下儿童后天性心脏病的主要病因,在中国、日本、韩国等东亚儿童中高发。如果治疗不及时,可能会在发热2-3周后发生冠状动脉瘤诱发的猝死。但川崎病的遗传免疫发生机制仍不清晰。
线粒体自噬是清除冗余或功能失调线粒体的重要途径。在线粒体极化丢失的情况, PINK1在线粒体外膜形成同源二聚体并发生自磷酸化,进而募集并磷酸化E3泛素连接酶Parkin。Parkin可使线粒体外膜蛋白泛素化,进而募集自噬适配器与LC3,启动线粒体自噬。在其他应激状态下,一些包含LC3-互作亚基(LIR)的线粒体蛋白,如NIX和FUNDC1,会通过直接与LC3结合诱导自噬。DRP1通过介导线粒体不对称裂变在Parkin非依赖的自噬中发挥关键作用。在生理或稳态情况下,线粒体自噬途径之间存在功能补偿,但在特定病理情况下,特定种类的线粒体自噬可能发挥主导作用。
在线粒体自噬缺陷的情况下,损伤线粒体来源的活性氧(ROS)和线粒体DNA(mtDNA)在心血管疾病的发生发展中发挥重要因素。ROS可使一氧化氮(NO)失活生成活性氮(RNS),NO缺乏会诱导血管平滑肌收缩、炎症浸润和血小板激活。mtDNA则可以通过TLR9和cGAS-STING激活炎症和IFN-I通路。cGAS-STING通路的过度激活促进了心血管疾病,如动脉粥样硬化、主动脉瘤和主动脉夹层等的发病。
2023年8月11日,来自广州市妇女儿童医疗中心的张玉霞和顾晓琼教授团队和来自重庆国际免疫中心的吴玉章教授团队联合在Nature Cardiovascular Research杂志上在线发表题“MCM8 mediated mitophagy protects vascular health in response to nitric oxide signaling in a mouse model of Kawasaki disease”的研究文章。该研究发现染色体维持复合体8蛋白(MCM8)的表达在并发冠状动脉瘤的川崎病患儿中显著下降。机制研究发现,NO促进MCM8和E3泛素连接酶TRIM21介导的线粒体自噬,维持冠状动脉的正常功能。而东亚人群常见的MCM8-P276变异介导的线粒体自噬能力下降,导致cGAS-STING-I型干扰素(IFN-I)通路过度激活,或与川崎病的易感性相关。

本研究通过构建Mcm8-/-小鼠,发现MCM8缺失可自发诱导冠状动脉增厚。在乳酸杆菌细胞壁提取物(LCWE)诱导的川崎病模型中,Mcm8-/-小鼠冠状动脉炎的严重程度较野生型小鼠显著加剧。RNA-seq分析显示,LCWE处理的Mcm8-/-小鼠心脏组织的mRNA表达谱呈现IFN-I信号通路激活以及心脏衰竭的特征。与之对应,川崎病患者血清中IFN-α2和IFN-β的表达水平相较于健康对照和普通发热患者显著升高。对比健康儿童,并发冠状动脉瘤的川崎病患儿外周血单核细胞MCM8表达显著降低,而cGAS和干扰素诱导基因(ISG)则显著上调(图1)。
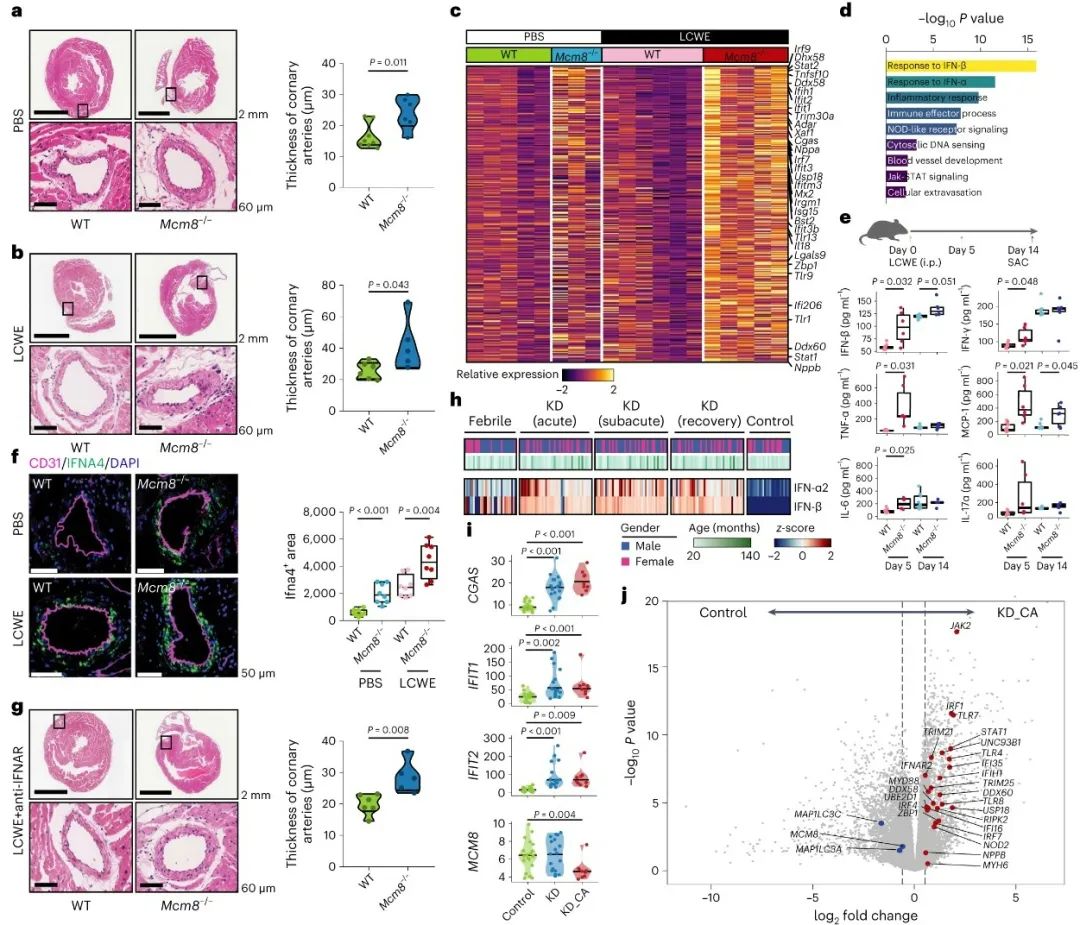
图1. 在LCWE诱导的川崎病模型中MCM8缺失促进IFN-I表达和冠状动脉血管病变
为了探究MCM8缺乏促进IFN-I表达的机制,研究者通过siRNA敲低了人单核细胞系(THP-1)、人脐静脉内皮细胞(HUVECs)中的MCM8或从Mcm8-/-小鼠中分离培养了骨髓源性的巨噬细胞(BMDM)和胚胎成纤维细胞(MEF),证实敲低或敲除MCM8能够促进IFN-I的表达和IRF3的磷酸化。通过敲除IFN-I信号上游的DNA和RNA感受器及信号传导蛋白,发现MCM8缺失通过cGAS-STING-IRF3激活了IFN-I信号通路。通过免疫共沉淀和质谱的方法,发现MCM8与BAX、BAK和VDAC1存在相互作用,且相互作用在细胞处于激活状态下明显明显增强。通过对线粒体膜电势的染色(TMRM),发现MCM8缺陷细胞在细胞应激状态下存在更多去极化损伤线粒体。基于以上发现,研究者推测mtDNA的累积可能是由于自噬缺陷导致的损伤线粒体累积造成的。研究者将Hela细胞的MCM8敲低,共聚焦显微镜分析显示MCM8缺失显著降低了LC3与线粒体的共定位。研究者进一步研究发现,MCM8存在LIR亚基,可以作为一个自噬受体与LC3结合。有意思的是,东亚人群特异的变异位点P276刚好位于MCM8的LIR结构域,该变异抑制了MCM8与LC3的结合,抑制了线粒体自噬(图2)。

图2. MCM8存在LIR结构域,作为自噬受体与LC3结合
以往研究认为,MCM8与MCM9形成复合体,主要定位于细胞核,调节基因组复制及损伤修复。临床上,与MCM8或MCM9相关的隐性复合杂合突变,或Mcm8-/-以及Mcm9-/-的小鼠主要表现为卵巢早衰以及配子发生障碍。那么,MCM8/MCM9相关的基因组损伤修复机制是否调节MCM8介导的线粒体自噬?通过敲减MCM9以及基因组损伤修复关键基因ATM,研究者发现MCM8介导的线粒体自噬不依赖于其参与的基因组损伤修复功能(图3)。
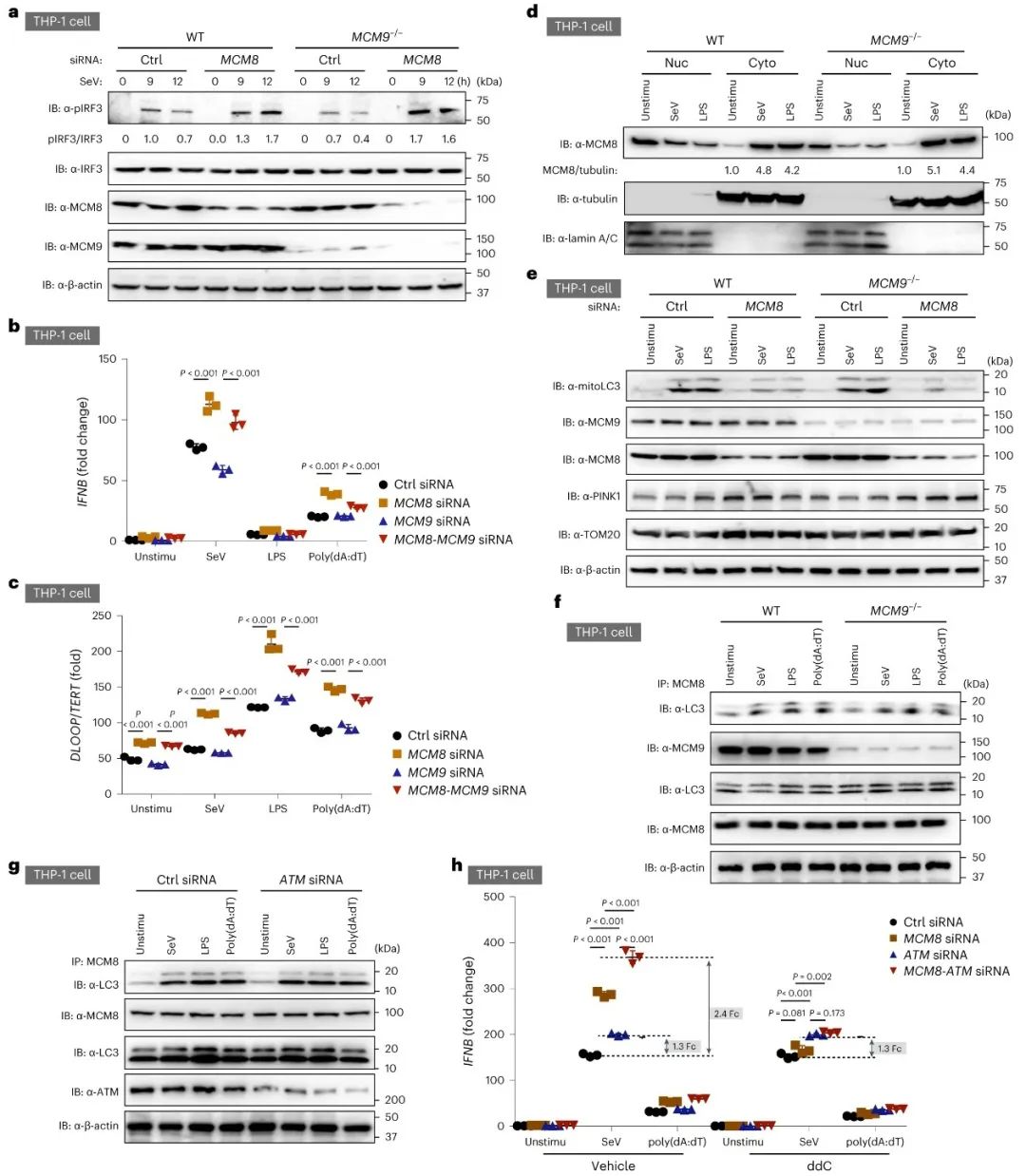
图3. MCM8介导的自噬不依赖于基因组损伤修复反应
那么,MCM8介导的线粒体自噬是否依赖于PINK1和PARKIN介导的线粒体泛素化途径或者DRP1介导的线粒体不对称裂变?通过敲减PINK1、PARKIN、FUNDC1、NIX以及DRP1,研究者发现MCM8介导的线粒体自噬不依赖于先前报道的线粒体自噬机制(图4)。

图4. MCM8介导的线粒体自噬不依赖于PINK1、PRKN、NIX、FUNDC1和DRP1
MCM8作为核蛋白,如何进入细胞质并启动线粒体自噬?通过核质分离实验,研究者发现MCM8在刺激情况下可以出核,而通过leptomycin B抑制核孔运输蛋白Exportin1后,MCM8的出核和介导自噬的功能均受到抑制。由于MCM8缺乏核输出信号,接下来研究者探索了泛素化是否参与了MCM8的出核。通过分析免疫共沉淀和质谱结果,研究者发现TRIM21可以结合MCM8并泛素化MCM8-MCM9结合界面的赖氨酸(K208、K232),诱导MCM8-MCM9复合体的解离。另外,K208位点的泛素化也促进了MCM8的出核。TRIM21主要定位于细胞质,但在NO信号激活后,可以进入细胞核。通过一系列细胞及动物试验,研究者证实清除NO(如利用Nos2-/-小鼠以及1400 W螯合剂)或者敲减TRIM21均抑制了MCM8的泛素化、出核以及介导线粒体自噬(图5)。
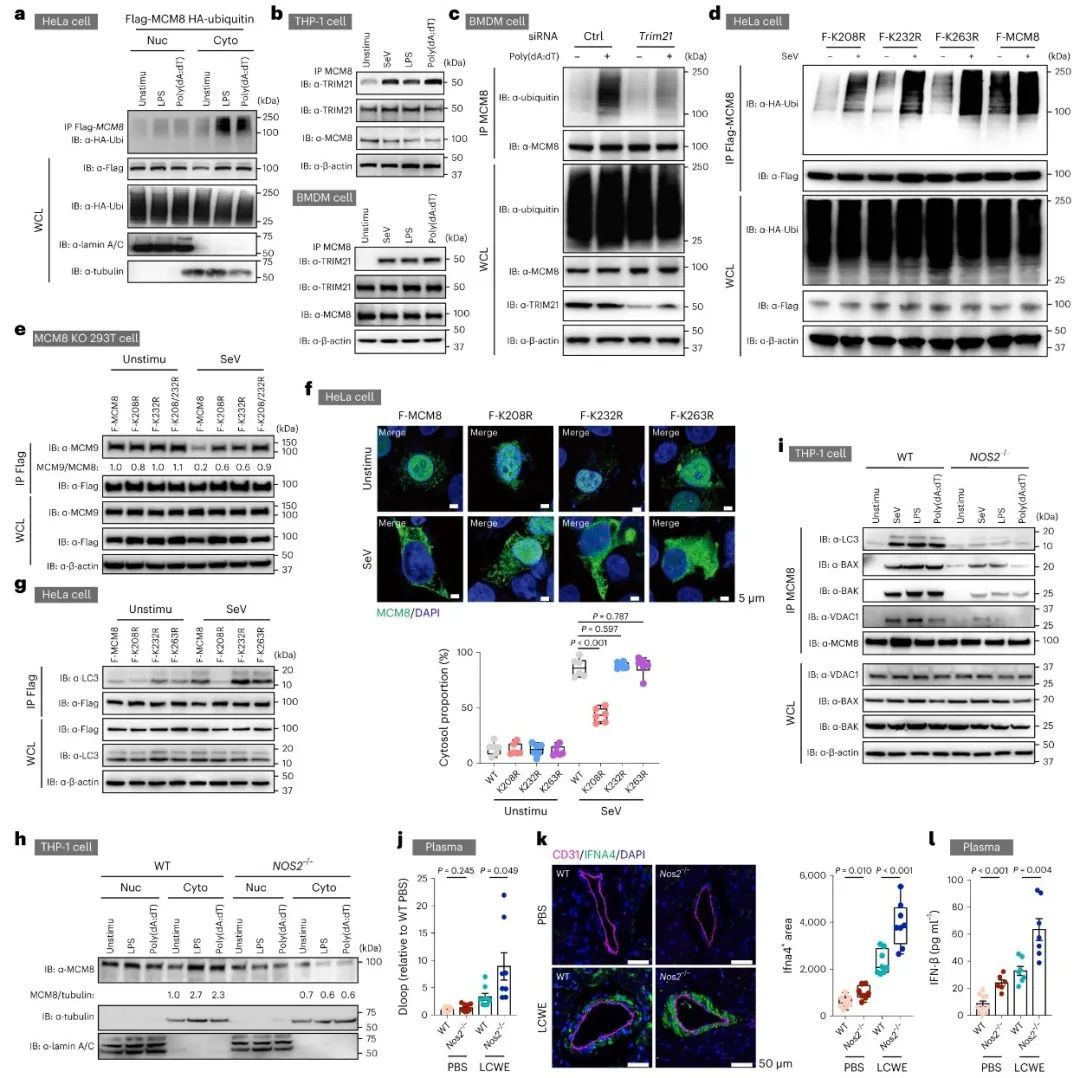
图5. NO信号触发TRIM21介导的MCM8泛素化、出核和诱导线粒体自噬
为了进一步证实东亚人群特异的MCM8-P276变异是否具有致病性,研究者对来自野生型(MCM8-A276)以及变异型(MCM8-P276)的人B细胞进行了永生化,发现MCM8-P276变异介导的线粒体自噬机制受损,导致mtDNA-cGAS-STING-IFN-I通路激活。另外,通过过继回输转染了人MCM8-A276或者人MCM8-P276基因的Mcm8-/-小鼠骨髓细胞,研究者发现MCM8-P276可以在LCWE刺激下诱导更为严重的冠状动脉损伤及心脏病理。总之,该研究报道了MCM8缺失诱发川崎患儿合并冠状动脉瘤的新机制,即NO信号通路促进TRIM21介导的MCM8泛素化,破环MCM8-MCM9复合体的稳定性,促进MCM8出核。在胞质,MCM8重新定位到线粒体孔道蛋白(BAX/BAK、VDAC1)并通过TRIM21促进它们的泛素化。此外,MCM8通过其自身的LIR亚基直接募集LC3并启动线粒体自噬(图6)。
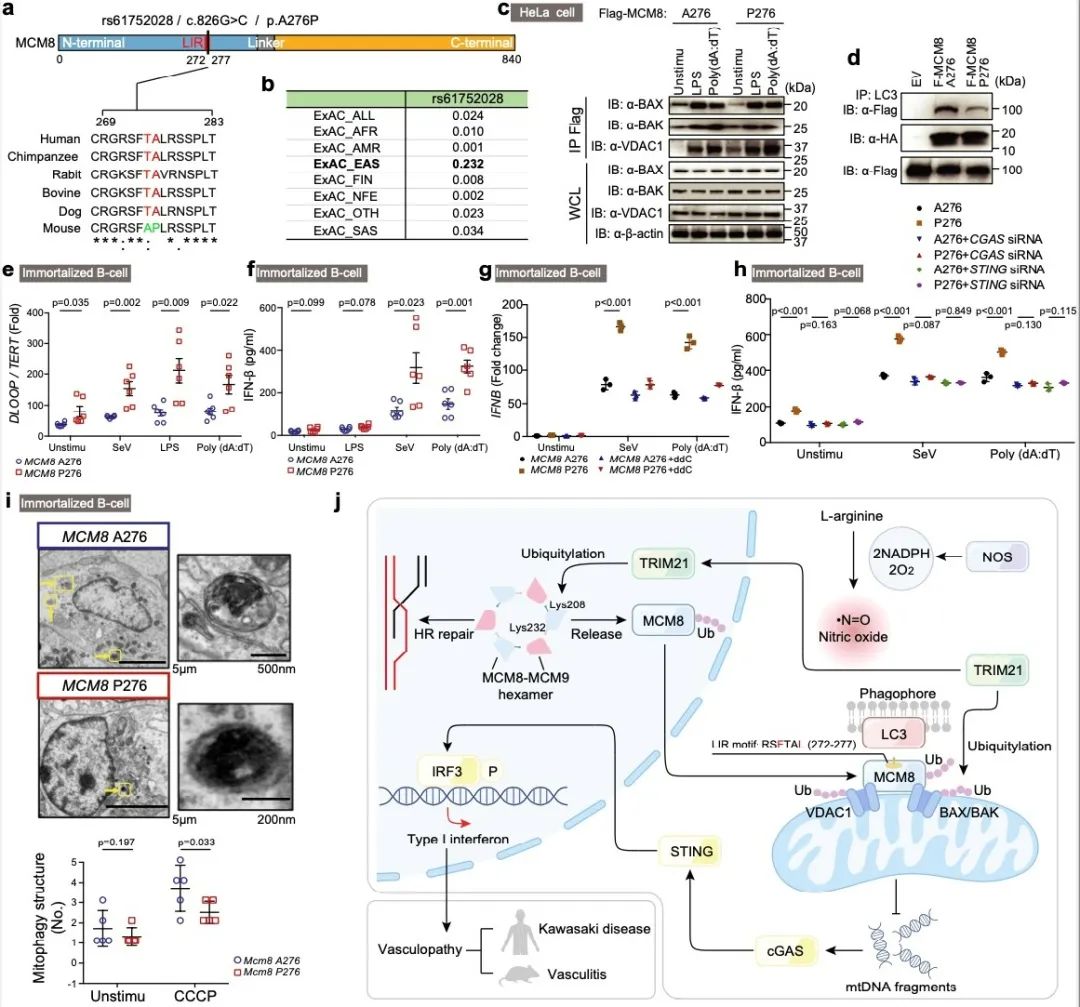
图6. MCM8介导的线粒体自噬机制及MCM8-P276变异的致病机理
NO是一个广泛存在的信号分子,在心血管、代谢、神经、免疫等多种生理病理过程中发挥关键调节作用。一氧化氮信号通路异常毫无意外也是许多主要疾病如心血管、炎症、免疫、神经、消化、代谢和肿瘤等疾病的核心特征。NO主要由NO合成酶(iNOS、eNOS、nNOS)催化L-精氨酸、O2和NADPH而来。剪切力、炎症细胞因子、细胞内钙离子流、多种激酶(AMPK、PKA、AKT)以及氧化还原状态均可以调节NO的供应。这些过程是否也影响MCM8介导的自噬,进而影响心血管相关生理病理以及其他疾病的发生发展仍需进一步深入研究。另外,是否可以通过恢复MCM8介导的线粒体自噬或阻断下游的IFN-1-JAK-STAT通路治疗包括川崎病在内的心血管及其它疾病也值得探索。
原文链接:
https://www.nature.com/articles/s44161-023-00314-x

