npj Precis Oncol:多中心研究助力肿瘤全外显子检测(WES)的临床标准化
时间:2023-11-23 11:24:43 热度:37.1℃ 作者:网络
随着医学的进步,人们发现很多癌症即使属于同一类型也可能因为异质性分为不同亚型。通常,对于癌症患者,医生首先要进行诊断和分期,然后选择合适的治疗手段。在医疗研究中,对于精准肿瘤学的呼声越来越高。从研究成果上看,微卫星不稳定性(MSI)、肿瘤突变负荷(TMB)和同源重组缺陷(HRD)等已被证明是对患者分层有意义的预测生物标志物。基因测序在临床的应用,已从包含几个到100个基因的较小panel转向约1Mb的大panel。这一转变表明,在临床实践中,加入基因测序作为癌症诊疗常规手段已经成为未来的趋势。
基于拷贝数改变(CNA)以及TMB、HRD和MSI复杂生物标志物的准确检测,全外显子组测序(WES)可提供对整个编码序列的无偏分析。但目前WES诊断主要用于部分研究领域,并未进入临床广泛性使用。原因之一是WES测序尚未经过充分的诊断稳定性验证(即在不同的医疗环境下,面对同样的病患,当按照标准流程进行检测后均可以得出一致性结果)。为了填补这一空白,德国个性化医疗中心(ZPM)开展了一项联合试点研究,以促进WES在常规癌症诊断中的使用。

来自德国海德堡大学研究团队和ZPM团队在Nature旗下的npj Precision Oncology期刊上发表研究文章“Multicentric pilot study to standardize clinical whole exome sequencing (WES) for cancer patients”。研究团队将30名患者的肿瘤和匹配正常DNA样本在四个ZPM实验室进行了WES,随后在五个ZPM生物信息机构进行了数据分析(图1),队列涉及20余种罕见的癌症类型,包括癌、肉瘤和其他肿瘤实体。结果显示,大多数突变被所有机构检测到(52%),几乎所有的临床相关突变(16/17)被所有机构一致地识别出来,体细胞突变检测高度一致,阳性百分比一致(PPA)为91%~95%,阳性预测值(PPV)为82%~95%。CNAs显示76%的基因组序列与高湿实验室可变性完全一致。复杂生物标志物检测在机构之间有很强的相关性(HRD为0.79-1,TMB为0.97-0.99),所有机构的MSI检测结果一致。
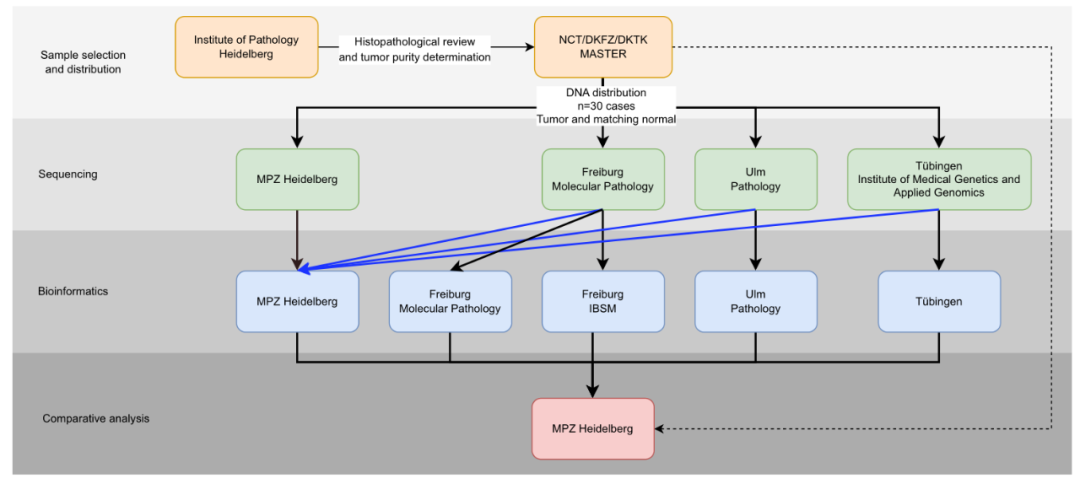
图1. 研究的试验设计。
一、关于体细胞突变
研究人员根据染色体位置和DNA水平的改变,对不同机构之间的体细胞突变进行了比较。只有完全匹配的才被认为是相同突变。五个参与机构总共进行了960个体细胞突变检测,包括804个单核苷酸变异(SNV)和156个删除/插入(Indel)。突变检测对应于270个独特突变,其中141个(52%)突变被所有机构检测到,59个(22%)独特突变由2-4个机构检测到,70个(26%)突变由单一机构检测到(图2)。
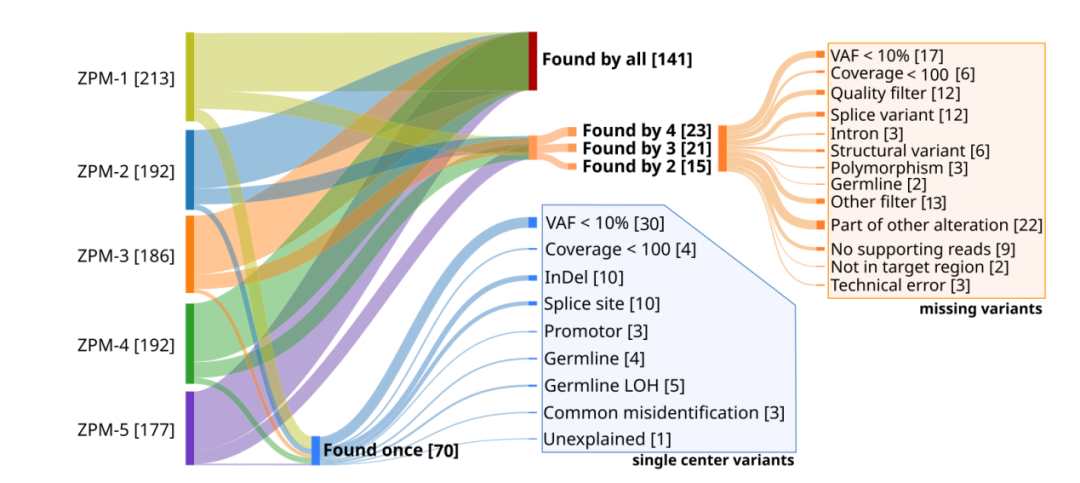
图2. 检测到的体细胞突变的机构间一致性。
研究团队分别分析了被2-4个机构检测到但被其他机构遗漏的突变(潜在的假阴性)和仅被一个机构检测到的突变(潜在的假阳性),对差异突变检测进行了深入分析。总的来说,59个独特突变被部分机构发现(23个突变被4个机构发现,21个突变被3个机构发现,15突变被2个机构发现),对应于110个遗漏的发现,其中23个(21%)的VAF<10%或覆盖率<100(图2,橙色)。只有一个机构检测到了70突变,其中30个(43%)突变的VAF<10%,4个(6%)突变的覆盖率< 100(图2,蓝色)。
研究团队根据3个不同的参考文献对参与机构的表现进行了评估。当使用3个机构共识作为参考时,在30个病例中,所有机构对9个病例的敏感性达到100%,对16个病例的敏感性至少达到85%。5家机构在3个机构参考、2个机构参考和TSO500数据参考方面的正百分比一致性(PPA)分别为91%~95%、87%~91%和90%~94%,相应的PPV分别为82%~95%、85%~98%和78%~86%(图3b)。在17种临床相关突变中,16种被所有五个机构检测到(图3c)。
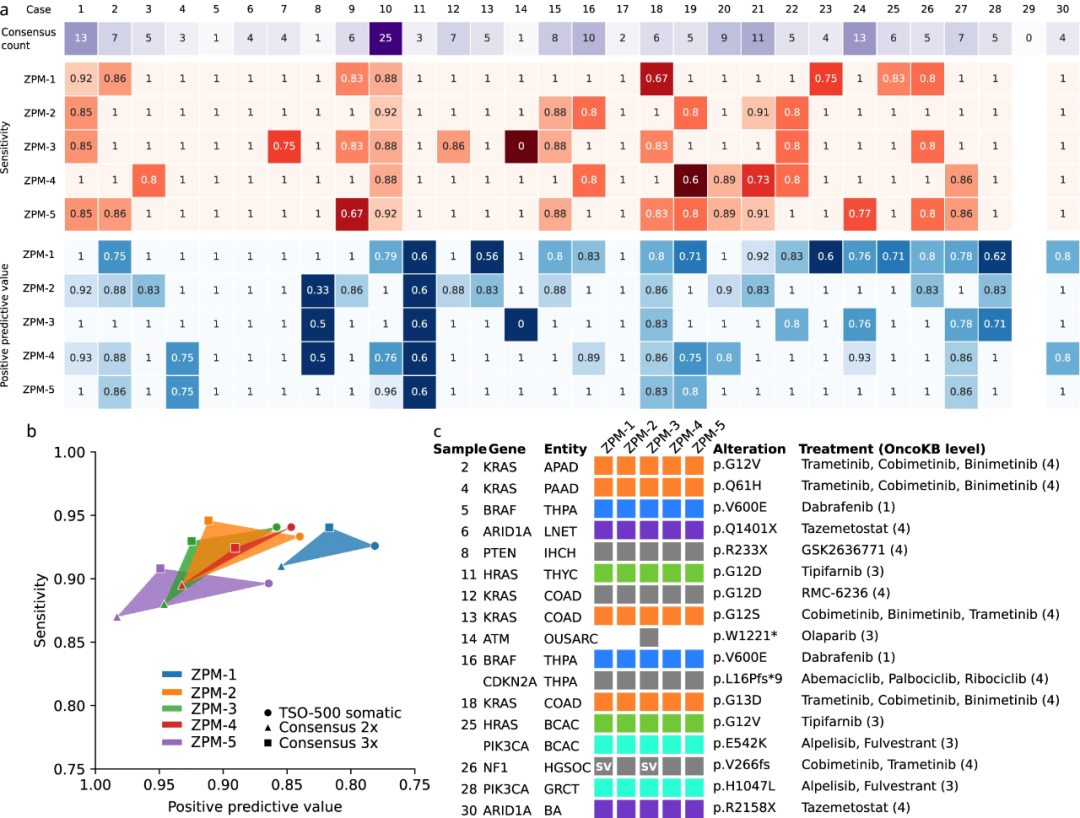
图3.将每个机构检测到的体细胞突变与包括至少3个机构检测到的所有突变的列表进行比较。
研究团队在机构之间成对比较了恒定拷贝数(CN))区域中的基因组片段和治疗相关基因扩增和缺失。在30个肿瘤结果中,76%的基因组区域完全匹配或考虑基因组复制调用后匹配(图4a,绿色/紫色条),24%的基因组区域拷贝数不匹配(图4a,红色条)。对于16个(73%)高度扩增的基因,所有机构都报告了高水平基因扩增。在5个基因中,只有2-4个机构报告了高水平扩增,其他机构报告了低于高水平阈值的扩增。对于剩余的单个基因,4个机构报告了高水平的扩增,1个机构没有报告任何扩增。所有机构检测到11个缺失(48%)。当使用统一的生物信息框架进行数据处理时,这种差异持续存在,意味着这是由湿实验室可变性引起的。(图4b)
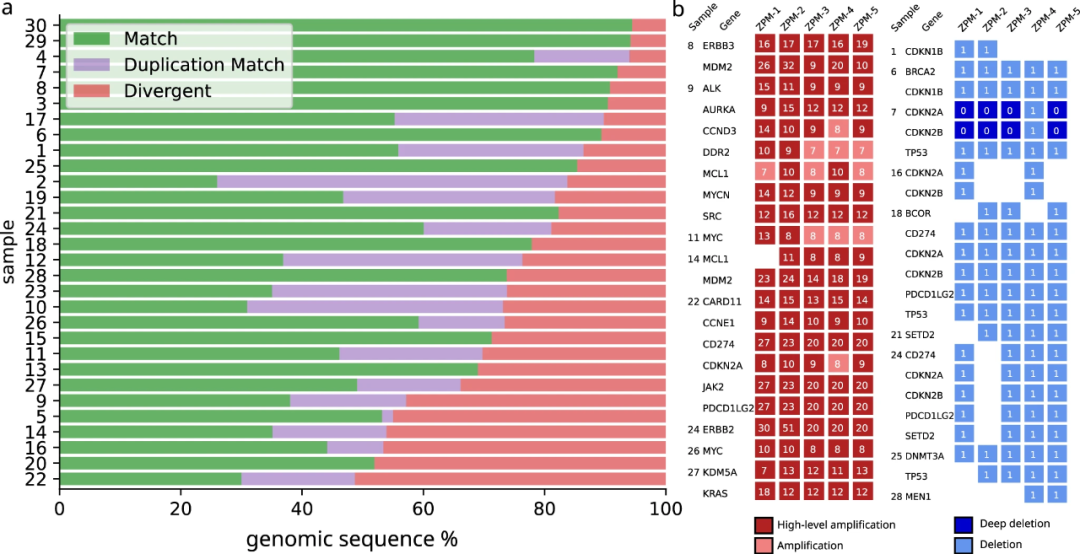
图4.CNA检测的机构间一致性。
二、关于复杂生物标志物
30个肿瘤的同源重组缺陷(HRD)评分在各机构之间具有很强的相关性(图3),机构之间的系统偏差在-15%到+9%的范围内。4个机构使用相同的生物信息学工作流程(Sequenza和scrapHRD)和相同的HRD调用截止点42,其中1个机构使用ClinCNV。检测到不同数量的HRR缺陷。30个肿瘤的TMB评分在各机构之间具有很强的相关性(图5)。深入分析显示,机构ZPM-5的TMB评分较ZPM-1、ZPM-2、ZPM-3和ZPM-4低9-20%,存在显著的系统偏差。所有机构均成功将两个高TMB病患和一个低TMB病患报告出来(图5)。
研究队列中的所有肿瘤均检测为MSS/MSI-L。因此,没有一个机构报告任何肿瘤的MSI-H(图5)。四家机构使用相同的生物信息学工具MSIsensor-pro,结果是MSI分数之间高度一致。ZPM-1使用Mantis工具虽然使MSI分数与其他机构的相关性较低,但MSI状态保持一致。
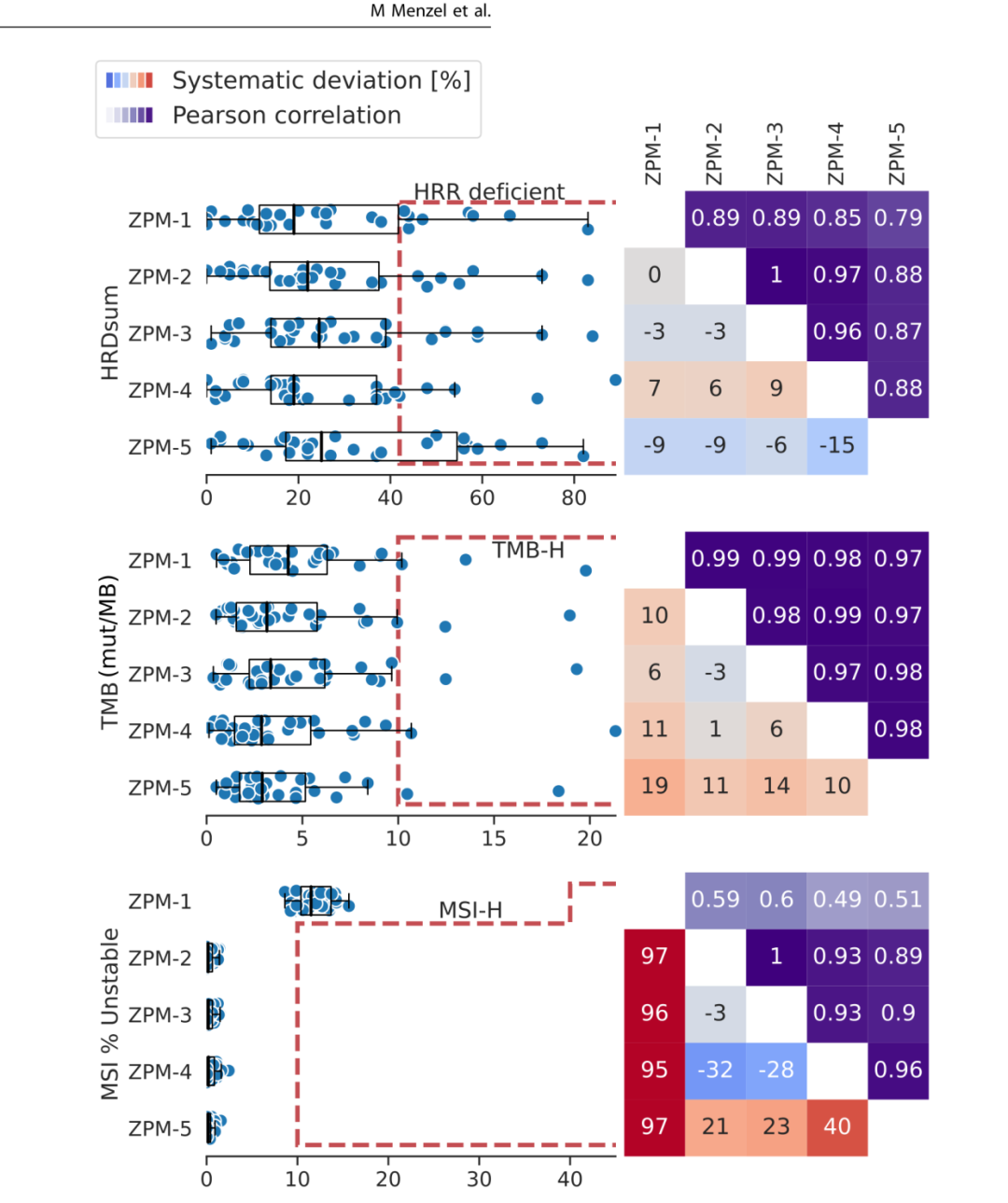
图5. HRD、TMB和MSI评分的机构间一致性。
三、剖析湿实验室和生物信息平台带来的差异
研究团队还区分了湿实验室和生物信息差异,发现湿实验室效应和生物信息学效应分离的中位标准差(SD)对每个生物标志物的影响相对较小(图6)。对于部分样本,可同时观察到多种复杂生物标志物的高变异性。此外,肿瘤纯度和倍性估计对HRDsum评分产生影响,需要对肿瘤纯度进行准确的组织学测定,以改进正确HRD评分的选择。
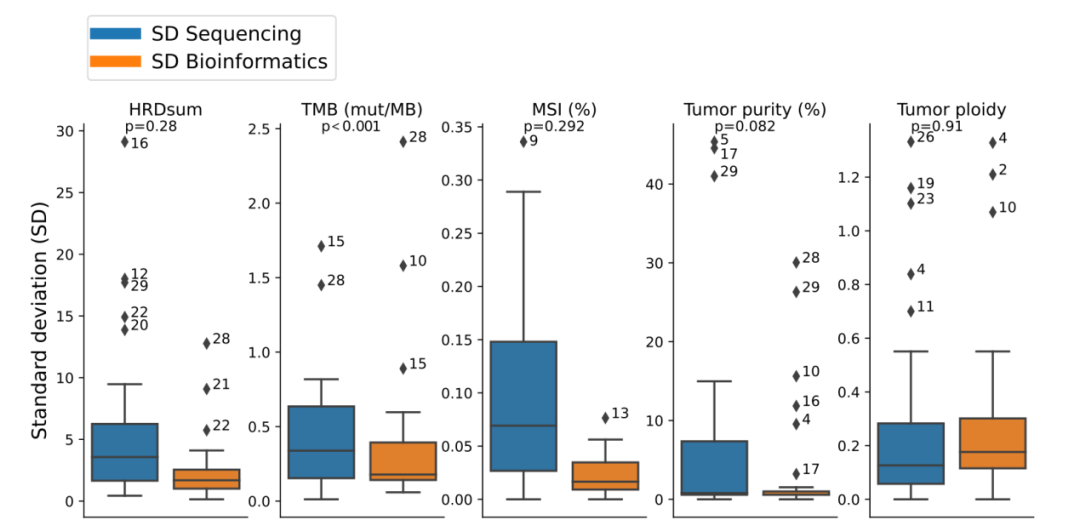
图6. 生物信息学和湿实验室机构间变异性的比较。
随着测序能力的提高和测序成本的降低,WES甚至WGS都有可能进入临床环境。与panel测序相比,WES可全面覆盖编码序列,可以根据当前和未来的诊断需求调整整个外显子组或虚拟基因panel;通过避免定制基因panel的偏差,促进不同实验室之间的可比性和标准化,因为定制基因panel可能在设计上不同,需要重复更新基因内容;准确检测(而不是估计)复杂生物标志物,例如TMB、HRD和MSI。
研究人员对临床癌症样本的WES分析进行了多机构的比较,涵盖了从样本到结果的工作流程,旨在评估一致性水平并确定机构间差异的因素。为此,研究人员分析了(1)结果的机构间一致性和(2)影响结果的干实验室和湿实验室因素。研究发现大多数突变被所有机构检测到(52%),几乎所有的临床相关突变(16/17)都被五个参与机构一致地识别出来。Indels(33%)比SNV(20%)更容易被遗漏,而假阳性突变检测在indels和SNV中是相似的。
总的来说,这些数据为WES作为临床级检测的标准化和协调提供了坚实的信息来源和基础,并有助于未来外部质量评估方案的概念化和设计。
论文原文:
Menzel, M., Ossowski, S., Kral, S., Metzger, P., Horak, P., Marienfeld, R., ... & Stenzinger, A. (2023). Multicentric pilot study to standardize clinical whole exome sequencing (WES) for cancer patients. NPJ Precision Oncology, 7(1), 106.
https://www.nature.com/articles/s41698-023-00457-x
