【综述】| NRAS突变型晚期黑色素瘤的治疗进展
时间:2023-12-13 14:38:18 热度:37.1℃ 作者:网络
[摘要] 黑色素瘤的发生、发展与多种癌基因的激活密切相关。15%~20%的黑色素瘤患者存在NRAS基因的激活突变,携带该突变基因的黑色素瘤具有更强的侵袭性,治疗难度大。由于RAS蛋白突变位点属于弱药物靶标,目前尚缺乏有效的靶向抑制剂,因此临床上多以免疫检查点抑制剂作为NRAS突变型晚期黑色素瘤的一线治疗方案,然而治疗反应率较低。近年来,在NRAS突变亚型中靶向治疗方案的探索主要集中在NRAS下游丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)通路,但结果不一:新型MEK1/2抑制剂tunlametinib,在晚期NRAS突变患者中总体客观缓解率(objective response rate,ORR)达到34.7%,较既往的binimetinib显著提高;然而泛RAF抑制剂belvarafenib和ERK抑制剂ulixertinib的Ⅰ期临床试验却未能展示出该药明显的优势。此外,以MEK抑制剂为基础的联合治疗也取得一定进展,现有证据表明,分子抑制剂类药物较免疫检查点抑制剂显示出更多的优势:选择性BRAF/CRAF抑制剂naporafenib(LXH254)与MEK抑制剂trametinib联合治疗NRAS突变型黑色素瘤的Ⅰb期临床试验中ORR达到46.7%;细胞周期蛋白依赖性激酶4/6(cyclin-dependent kinase4/6,CDK4/6)抑制剂ribociclib和binimetinib联合治疗在携带NRAS突变同时合并细胞周期蛋白基因异常的人群中可达32.5%;黏着斑激酶(focal adhesion kinase,FAK)抑制剂IN10018联合cobimetinib的研究结果也表现出较好的ORR(38.5%);但免疫检查点抑制剂程序性死亡[蛋白]配体-1(programmed death ligand-1,PD-L1)单抗durvalumab联合trametinib方案仅使27.2%的患者达到部分缓解(3/11)。与此同时,部分临床前研究结果也显示出一些转化潜质:如热激蛋白90(heat shock protein 90,HSP90)抑制剂XL888和丝氨酸/苏氨酸蛋白激酶19(serine/threonine protein kinase 19,STK19)抑制剂均在动物模型中表现出显著抑制NRAS突变型黑色素瘤细胞生长的能力。本文综述了NRAS突变型黑色素瘤的致癌机制及近年来治疗领域的研究进展,旨在展示该亚型患者的治疗现状,对多种新型治疗方法的临床研究结果进行总结和归纳,为当前临床实践和未来联合治疗方案提供依据。
[关键词] NRAS基因突变;黑色素瘤;致癌机制;靶向治疗
[Abstract] Oncogenic mutations are responsible for a majority of the malignancy of melanoma. Activating mutations of NRAS gene are found in 15%-20% melanoma cases, endowing the tumor cells with more aggressive phenotypes and greater difficulty to treat. The development of targeted inhibitor of mutant NRAS remains a big challenge since the mutation sites could hardly be druggable. Therefore, immune checkpoint inhibitors are currently recommended as the first-line therapy for NRAS mutant advanced melanoma albeit the response rate is still far from satisfaction. In recent years, the exploration of targeted therapy regimens has focused on the downstream pathway of NRAS, the mitogen-activated protein kinase (MAPK) pathway. A novel MEK1/2 inhibitor tunlametinib was reported to achieve an objective response rate (ORR) of 34.7% which is higher than the ORR of binimetinib in previous report. However, the phase Ⅰ trial of the pan-RAF inhibitor belvarafenib and the ERK inhibitor ulixertinib failed to show marked benefits. In the meanwhile, MEK inhibitor-based combination therapy has also achieved some progress: it was reported in the phase Ⅰb trial of the selective BRAF/CRAF inhibitor naporafenib (LXH254) combined with Trametinib in NRAS mutant melanoma that the ORR was 46.7%. The ORR of binetinib plus the cyclin-dependent kinase 4/6 (CDK4/6) inhibitor, ribociclib, was 32.5% in patients with NRAS mutation with concurrent alterations of CDKN2A, CDK4, or CCND1. The response rate of the combination of focal adhesion kinase (FAK) inhibitor, IN10018, and cobimetinib was 38.5%. On the other hand, only 27.2% of patients carrying NRAS mutation responded partially to the combined regimen of immune checkpoint inhibitor programmed death ligand-1 (PD-L1) monoclonal antibody durvalumab+trametinib. In addition, some preclinical findings have also shown translational potentials: for example, heat shock protein 90 (HSP90) inhibitor XL888 and serine/threonine protein kinase 19 (STK19) inhibitors were found to inhibit the growth of NRAS mutant melanoma in animal models. This article reviewed the oncogenic roles of NRAS mutation in melanoma and the cutting-edge clinical trials for the treatment of NRAS mutant melanoma, aiming to provide alterative treatment options for clinical practice and inspire novel combination regimen.
[Key words]NRAS mutation; Melanoma; Carcinogenesis; Targeted therapy
NRAS和BRAF是人类黑色素瘤中突变率最高的两个驱动基因,其中,针对BRAFV600位点突变的靶向抑制剂已获得较高的治疗反应率,但携带NRAS突变的病例治疗仍十分困难。RAS基因家族作为一类原癌基因,在进化中较为保守,参与细胞增殖、凋亡、分化和骨架构建等多种重要生物学过程,本文就NRAS突变型黑色素瘤治疗的新进展予以综述。
1 NRAS基因在黑色素瘤中的促癌机制
RAS基因家族包含3种功能性基因:NRAS、 KRAS和HRAS,分别位于1号、11号、12号染色体短臂上[1],编码产物为相对分子质量均为21×103的G蛋白,属于一种小GTP酶,经鸟嘌呤核苷酸交换因子(guanine nucleotide-exchange factor,GEF)介导活化,再由GTP酶活化蛋白(GTPase-activating protein,GAP)催化水解还原为无活性形式,以两种状态循环的方式进行活性调节[2],被称为细胞的“分子开关”。
高加索人群和中国人群的黑色素瘤NRAS突变率相近,分别为15.0%~20.0%[3]和10.9%~20.0%[4],多发生于第61位密码子,并以Q61R为主,突变后的细胞中NRAS处于持续激活状态,相比于BRAF/NRAS双野生甚至BRAF突变型病例,携带NRAS突变的黑色素瘤侵袭性更强[5-7]。在正常细胞中,RAS活化促进BRAF/CRAF形成二聚体,二聚后的RAF兄弟蛋白才被激活,与野生型细胞(由BRAF负责信号转导)不同,在NRAS突变的黑色素瘤细胞中,信号主要由CRAF转导[8]。
活化的RAS蛋白主要通过3条信号转导通路发挥对黑色素瘤的促癌作用[9](图1)。① MAPK信号转导通路:GTP结合的RAS变构激活RAF,随后依次级联激活丝裂原活化蛋白激酶激酶(MAPKK)家族(MEK1、MEK2)及MAPK家族(ERK1、ERK2),推动细胞周期、促进增殖[10-14]。② 磷脂酰肌醇3激酶(phosphatidylinositide3-kinases,PI3K)/丝氨酸-苏氨酸激酶(serine/threonine kinase proteins,AKT)(PI3K-AKT)信号转导通路[15]:PI3K由RAS激活后磷酸化磷脂酰肌醇-4,5-二磷酸[phosphatidylinositol(4,5)bisphosphate,PIP2]分子生成磷酸化磷脂酰肌醇-3,4,5-三磷酸[phosphatidylinositol(3,4,5)trisphosphate,PIP3],后者招募激活AKT。AKT可以通过磷酸化底物抑制TP53降解并抑制多种促凋亡蛋白活性[16],阻断凋亡。此外,AKT触发级联反应,导致哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)的激活,通过促进细胞代谢、推动细胞周期及抑制自噬等方式促进肿瘤增殖[17]。③ RAL信号转导通路[18]:RAL(RAS-like)GTPases属于RAS小GTP酶,包括Ral-鸟嘌呤核苷酸交换因子(Ral-GEF),例如Ral-GDS、RalA和RalB等,在活性GTP和非活性GDP结合构象之间循环。RalGEF的激活促进黑色素瘤的增殖[18]和锚定性生长[19-20];Ral还可以通过与Ral相互作用蛋白RLIP76(也称RalBP1)的结合,促进黑色素瘤血管的生成[22-22]。
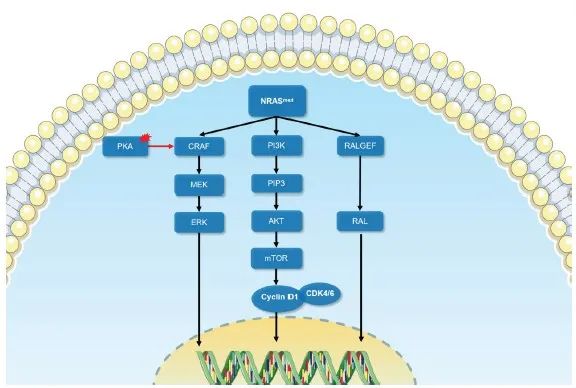
图1 NRAS突变的信号转导示意图
Fig. 1 The schematic diagram of signal transduction of NRAS mutation
Oncogenic signal transduction following NRAS mutation causes melanoma development mainly through MAPK signaling, PI3K-AKT signaling pathway and RAS-RAL signaling pathway. Black arrow: Conducted; Red arrow: Enhanced; Red area: Abnormal activation.
2 NRAS突变型黑色素瘤的治疗现状
由于缺乏有效的靶向抑制剂,目前美国国家综合癌症网络(National Comprehensive Cancer Network,NCCN)和欧洲肿瘤内科学会(European Society for Medical Oncology,ESMO)指南[23-24]中均推荐以免疫治疗作为NRAS突变型晚期黑色素瘤的一线治疗,但NRAS突变状态对免疫疗效的影响报道并不一致:Jaeger等[25]对16项临床研究进行系统评价发现NRAS突变的黑色素瘤病例比野生型对免疫检查点抑制剂具有更高的客观缓解率(objective response rate,ORR)(RR=1.28,95% CI:1.01~1.64)。然而在亚洲人群[26],接受程序性死亡[蛋白]-1(programmed death-1,PD-1)单抗治疗的NRAS突变型亚组疗效却显著差于野生型亚组:在皮肤型患者中,两亚组ORR分别为9.5%和23.9%;在非皮肤型患者中,两亚组ORR分别为0.0%和13.7%。少部分NRAS突变病例也可获益于MEK抑制剂:比美替尼(binimetinib)在NRAS突变型晚期患者的Ⅲ期临床试验(NEMO研究)证实,binimetinib组中位无进展生存期(median progression-free survival,mPFS)长于达卡巴嗪组(2.8个月 vs 1.5个月),但ORR仅为15%[27]。另一MEK抑制剂匹马赛替尼(pimasertib)在NRAS突变型患者中达到27%的ORR,虽高于达卡巴嗪对照组(14%),但中位总生存期(median overall survival,mOS)差异无统计学意义(HR=0.89,95% CI:0.61~1.30)[28]。由于临床获益有限,目前MEK抑制剂仅作为免疫治疗进展后的二线选择。
3 NRAS突变型黑色素瘤的治疗进展
3.1 单药治疗
由于突变位点已经明确,既往研究曾尝试针对突变位点的靶向治疗,然而不同于BRAF V600突变位点,RAS蛋白突变位点属于弱药物靶标,因为在核苷酸结合位点之外缺乏药物分子的结合口袋,GTP分子与RAS蛋白的亲和力达到皮摩级别,针对GTP本身的竞争性抑制药物已被证实无效[29]。其他的单药治疗策略包括以下几种:
3.1.1 RAS抑制剂
法尼基转移酶抑制剂(farnesyl transferase inhibitor,FTI):FTI的作用机制是通过抑制法尼基转移酶将法尼基结合到p21RAS羧基端的半胱氨酸(cysteine,Cys)残基,使其不能被羧甲基化,p21RAS就不能定位在细胞膜上,从而阻滞RAS的激活。但目前FTI阻断RAS翻译后修饰和细胞膜定位只能在理论上实现,一项FTI的Ⅱ 期临床试验入组的14例黑色素瘤患者均无治疗反应,已提前终止[30]。此外,此类药物显著的毒性也限制了其进一步开发及临床转化[31-32]。
新近公布KRAS抑制剂索托拉西布(sotorasib,KRAS G12C靶向抑制剂,NCT03600883)可明显延长KRAS G12C突变实体瘤患者的mPFS[33-34],这一结果提示该药或将给携带NRAS G12C突变的黑色素瘤患者带来获益,但仍需临床数据验证。
3.1.2 RAF-MAPK通路抑制剂
由于针对NRAS突变位点的靶向抑制剂开发进展缓慢,因此NRAS突变型黑色素瘤的治疗策略主要是通过抑制其活化后的下游效应,目前主要集中在RAF-MAPK通路抑制剂。
3.1.2.1 RAF抑制剂
经shRNA同时敲低BRAF和CRAF可显著抑制NRAS突变型黑色素瘤的生长[35],因此,泛RAF抑制剂或可有效地阻断NRAS信号向MEK的传递。在泛RAF抑制剂belvarafenib(又称RG6185或HM95573)的Ⅰ期临床试验中其ORR为11%, mPFS为25周[36]。
3.1.2.2 MEK1/2抑制剂
2023年美国临床肿瘤学会(American Society of Clinical Oncology,ASCO)会议上公布新型MEK1/2选择性抑制剂妥拉美替尼(tunlametinib、 HL-085)在晚期NRAS突变型黑色素瘤的疗效数据显示总体ORR为34.7%,即使在免疫治疗失败的患者中,ORR也高达39.1%[37],较既往binimetinib的疗效有明显提高[27]。
3.1.2.3 ERK抑制剂
ERK是MAPK通路中的关键节点,抑制ERK活性可以提高NRAS突变黑色素瘤对MEK抑制剂的反应[38]。在NRAS突变黑色素细胞中,ERK抑制剂VX-11e对细胞增殖的抑制作用比MEK抑制剂更明显[39]。但在Ⅰ期临床试验中,应用ERK1/2抑制剂优立替尼(ulixertinib),仅13.5%的患者达到部分缓解[40]。
3.2 联合治疗
由于NRAS活化后激活的下游通路较多,单一通路抑制剂易产生耐药,故联合不同通路治疗也被不断尝试,主要策略是以MEK抑制剂为基础联合其他通路抑制剂或免疫治疗药物。
3.2.1 基于MEK抑制剂的联合方案
3.2.1.1 靶向抑制剂
NRAS突变型黑色素瘤细胞对泛RAF抑制剂的耐药与MAPK活化的相关性已被证实,故联合曲美替尼(trametinib)可增强泛RAF抑制剂的疗效[41]。目前正在进行的泛RAF抑制剂联合MEK抑制剂的临床试验有NCT02974725、NCT03284502等,NCT03284502目前公布的ORR为40%(n=9)。此外,选择性BRAF/CRAF抑制剂naporafenib(LXH254)与trametinib联合治疗KRAS/BRAF突变的非小细胞肺癌和NRAS突变黑色素瘤的Ⅰb期临床试验结果[42]显示,接受naporafenib 200 mg每日两次联合trametinib 1 mg每日2次治疗的患者的ORR和mPFS分别为46.7%和5.52个月。除了MAPK通路外,同时抑制PI3K-AKT通路也可产生协同作用[43]。PI3Kα抑制剂(alpelisib)联合binimetinib的Ⅰb期临床试验中ORR为20%[44]。而AKT抑制剂(GSK2141795)和trametinib的联合方案的Ⅱ期临床试验显示mPFS和mOS仅为2.3和4.0个月,ORR为0%,未能产生临床获益[45](表1)。
3.2.1.2 CDK4/6抑制剂
CDK4/6抑制剂瑞博西尼(ribociclib)和binimetinib联合治疗的Ⅱ期队列(n=41)结果显示,mPFS为3.7个月,总体ORR为19.5%,在伴有CDKN2A、CDK4或CCND1基因表达异常的患者表现出更高的ORR(32.5%),因此,携带NRAS突变和细胞周期基因异常的患者从该联合方案获益的可能性更高[46]。
3.2.1.3 FAK抑制剂
FAK抑制剂(IN10018)联合考比替尼(cobimetinib)在NRAS突变黑色素瘤患者的Ⅰ期临床试验中ORR为38.5%,mPFS为5.45个月[47]。
3.2.1.4 自噬抑制剂
已证实细胞自噬可保护肿瘤细胞免受RAS抑制剂的细胞毒性作用[48],现有临床试验(NCT03979651)正在评估自噬抑制剂(羟氯喹)联合MEK抑制剂治疗NRAS突变黑色素瘤的疗效,但研究结果尚未公布。
3.2.1.5 免疫检查点抑制剂
针对PD-L1的度伐利尤单抗(durvalumab)联合trametinib(同用或序贯)方案在NRAS突变患者中获得27.7%的部分缓解(3/11)[49]。IMspire170研究对比了cobimetinib联合PD-L1单抗阿替利珠单抗(atelizumab)和帕博利珠单抗(pembrolizumab)一线治疗BRAF野生型患者,发现两种治疗的mPFS(5.7个月 vs 5.5个月)及ORR(53% vs 65%)差异无统计学意义,即使在NRAS突变的患者中,cobimetinib联合atelizumab方案也未能显示出优势[50]。
3.2.2 基于酪氨酸激酶抑制剂的联合方案
NRAS突变黑色素瘤表达大量细胞表面受体酪氨酸激酶(receptor tyrosine kinase,RTK),具有驱动细胞增殖的潜力,例如Axl、ERBB2、c-MET、EGFR等[51-52]。索拉非尼(sorafenib,VEGFR抑制剂)联合MET酪氨酸激酶抑制剂(tivantinib)治疗晚期实体肿瘤的Ⅰ期临床试验中[53],NRAS突变黑色素瘤患者的ORR虽然低于野生型或未知状态的患者(20.0% vs 33.3%),但其mPFS却更长(5.4个月 vs 3.3个月),初步显示出TKI联合METi的协同疗效。


4 总结与展望
本综述所阐述的临床研究的具体情况见表1。目前针对NRAS突变型黑色素瘤的靶向和免疫治疗方案均未能媲美BRAF抑制剂在BRAF突变型病例中达到的治疗反应率,但近年来基于MEK抑制剂的部分联合治疗策略已经取得喜人的进展[54]。此外,临床前研究中,热激蛋白90(heat shock protein 90,HSP90)抑制剂[55]及丝氨酸/苏氨酸蛋白激酶19(serine/threonine protein kinase 19,STK19)抑制剂[56]也显示出很强的转化潜力,有望成为未来NRAS突变型黑色素瘤治疗的 突破口。
利益冲突声明:所有作者均声明不存在利益冲突。
[参考文献]
[1]TSAI F D, LOPES M S, ZHOU M, et al. KRAS4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif[J]. Proc Natl Acad Sci U S A, 2015, 112(3): 779-784.
[2]MACARA I G. The ras superfamily of molecular switches[J]. Cell Signal, 1991, 3(3): 179-187.
[3]JAKOB J A, BASSETT R L Jr, NG C S, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma[J]. Cancer, 2012, 118(16): 4014-4023.
[4]曾 颖, 康晓静, 张祥月, 等. 肢端型黑素瘤NRAS基因突变检测及预后分析[J]. 中华皮肤科杂志, 2016, 49(7): 474-477.
[5]LALLY S E, MILMAN T, ORLOFF M, et al. Mutational landscape and outcomes of conjunctival melanoma in 101 patients[J]. Ophthalmology, 2022, 129(6): 679-693.
[6]LISZKAY G, MÁTRAI Z, CZIRBESZ K, et al. Predictive and prognostic value of BRAF and NRAS mutation of 159 sentinel lymph node cases in melanoma-a retrospective single-institute study[J]. Cancers (Basel), 2021, 13(13): 3302.
[7]ZABLOCKA T, KREISMANE M, PJANOVA D, et al. Effects of BRAF V600E and NRAS mutational status on the progression-free survival and clinicopathological characteristics of patients with melanoma[J]. Oncol Lett, 2023, 25(1): 27.
[8]DUMAZ N, HAYWARD R, MARTIN J, et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling[J]. Cancer Res, 2006, 66(19): 9483-9491.
[9]PHADKE M S, SMALLEY K S M. Targeting NRAS mutations in advanced melanoma[J]. J Clin Oncol, 2023, 41(14): 2661-2664.
[10]SMALLEY K S, HAASS N K, BRAFFORD P A, et al. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases[J]. Mol Cancer Ther, 2006, 5(5): 1136-1144.
[11]SHARMA A, TRIVEDI N R, ZIMMERMAN M A, et al. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors[J]. Cancer Res, 2005, 65(6): 2412-2421.
[12]SATYAMOORTHY K, LI G, GERRERO M R, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation[J]. Cancer Res, 2003, 63(4): 756-759.
[13]ZHUANG L, LEE C S, SCOLYER R A, et al. Activation of the extracellular signal regulated kinase (ERK) pathway in human melanoma[J]. J Clin Pathol, 2005, 58(11): 1163-1169.
[14]OBA J, NAKAHARA T, ABE T, et al. Expression of c-Kit, p-ERK and cyclin D1 in malignant melanoma: an immunohistochemical study and analysis of prognostic value[J]. J Dermatol Sci, 2011, 62(2): 116-123.
[15]CANCER GENOME ATLAS NETWORK. Genomic classification of cutaneous melanoma[J]. Cell, 2015, 161(7): 1681-1696.
[16]ROBERTSON G P. Functional and therapeutic significance of Akt deregulation in malignant melanoma[J]. Cancer Metastasis Rev, 2005, 24(2): 273-285.
[17]XIE X Q, WHITE E P, MEHNERT J M. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma[J]. PLoS One, 2013, 8(1): e55096.
[18]ZIPFEL P A, BRADY D C, KASHATUS D F, et al. Ral activation promotes melanomagenesis[J]. Oncogene, 2010, 29(34): 4859-4864.
[19] MISHRA P J, HA L, RIEKER J, et al. Disp of RAS downstream pathways in melanomagenesis: a role for Ral in transformation[J]. Oncogene, 2010, 29(16): 2449-2456.
[20] FALSETTI S C, WANG D A, PENG H R, et al. Geranylgeranyltransferase Ⅰ inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth[J]. Mol Cell Biol, 2007, 27(22): 8003-8014.
[21] SINGHAL S S, AWASTHI Y C, AWASTHI S. Regression of melanoma in a murine model by RLIP76 depletion[J].
Cancer Res, 2006, 66(4): 2354-2360.
[22] MARTÍN M T, ALCALDE M, PLOU F J, et al. Covalent immobilization of cyclodextrin glucosyltransferase (CGTase) in activated silica and Sepharose[J]. Indian J Biochem Biophys, 2002, 39(4): 229-234.
[23] SWETTER S M, THOMPSON J A, ALBERTINI M R, et al. NCCN guidelines® insights: melanoma: cutaneous, version 2.2021[J]. J Natl Compr Canc Netw, 2021, 19(4): 364-376.
[24] MICHIELIN O, VAN AKKOOI A C J, ASCIERTO P A, et al. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up[J]. Ann Oncol, 2019, 30(12): 1884-1901.
[25] JAEGER Z J, RAVAL N S, MAVERAKIS N K A, et al. Objective response to immune checkpoint inhibitor therapy in NRAS-mutant melanoma: a systematic review and metaanalysis[J]. Front Med (Lausanne), 2023, 10: 1090737.
[26] ZHOU L, WANG X, CHI Z H, et al. Association of NRAS mutation with clinical outcomes of anti-PD-1 monotherapy in advanced melanoma: a pooled analysis of four Asian clinical trials[J]. Front Immunol, 2021, 12: 691032.
[27] DUMMER R, SCHADENDORF D, ASCIERTO P A, et al. Binimetinib versus dacarbazine in patients with advanced
NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial[J]. Lancet Oncol, 2017, 18(4):
435-445.
[28] LEBBÉ C, DUTRIAUX C, LESIMPLE T, et al. Pimasertib versus dacarbazine in patients with unresectable NRAS-mutated cutaneous melanoma: phase Ⅱ, randomized, controlled trial with crossover[J]. Cancers (Basel), 2020, 12(7): 1727.
[29] RYAN M B, CORCORAN R B. Therapeutic strategies to target RAS-mutant cancers[J]. Nat Rev Clin Oncol, 2018, 15(11): 709-720.
[30] GAJEWSKI T F, SALAMA A K S, NIEDZWIECKI D, et al. Phase Ⅱ study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104)[J]. J Transl Med, 2012, 10: 246.
[31] FEDORENKO I V, GIBNEY G T, SMALLEY K M. NRAS mutant melanoma: biological behavior and future strategies for therapeutic management[J]. Oncogene, 2013, 32(25): 3009-3018.
[32] TATEISHI K, TSUBAKI M, TAKEDA T, et al. FTI-277 and GGTI-289 induce apoptosis via inhibition of the Ras/ERK and Ras/mTOR pathway in head and neck carcinoma HEp-2 and HSC-3 cells[J]. J BUON, 2021, 26(2): 606-612.
[33] SKOULIDIS F, LI B T, DY G K, et al. Sotorasib for lung cancers with KRAS p.G12C mutation[J]. N Engl J Med, 2021, 384(25): 2371-2381.
[34] HONG D S, FAKIH M G, STRICKLER J H, et al. KRASG12C inhibition with sotorasib in advanced solid tumors[J]. N Engl J Med, 2020, 383(13): 1207-1217.
[35] JAISWAL B S, JANAKIRAMAN V, KLJAVIN N M, et al. Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors[J]. PLoS One, 2009, 4(5): e5717.
[36] YEN I, SHANAHAN F, LEE J, et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma[J]. Nature, 2021, 594(7863): 418-423.
[37] WANG X, LUO Z G, CHEN J, et al. First-in-human phase I dose-escalation and dose-expansion trial of the selective MEK inhibitor HL-085 in patients with advanced melanoma harboring NRAS mutations[J]. BMC Med, 2023, 21(1): 2.
[38] ADAM C, FUSI L, WEISS N, et al. Efficient suppression of NRAS-driven melanoma by Co-inhibition of ERK1/2 and
ERK5 MAPK pathways[J]. J Invest Dermatol, 2020, 140(12): 2455-2465.e10.
[39] CARLINO M S, TODD J R, GOWRISHANKAR K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma[J]. Mol Oncol, 2014, 8(3): 544-554.
[40] MENDZELEVSKI B, FERBER G, JANKU F, et al. Effect of ulixertinib, a novel ERK1/2 inhibitor, on the QT/QTc interval in patients with advanced solid tumor malignancies[J]. Cancer Chemother Pharmacol, 2018, 81(6): 1129-1141.
[41] ATEFI M, TITZ B, AVRAMIS E, et al. Combination of pan- RAF and MEK inhibitors in NRAS mutant melanoma[J]. Mol Cancer, 2015, 14(1): 27.
[42] DE BRAUD F, DOOMS C, HEIST R S, et al. Initial evidence for the efficacy of naporafenib in combination with trametinib in NRAS-mutant melanoma: results from the expansion arm of a phase ib, open-label study[J]. J Clin Oncol, 2023, 41(14): 2651-2660.
[43] POSCH C, MOSLEHI H, FEENEY L, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo[J]. Proc Natl Acad Sci U S A, 2013, 110(10): 4015-4020.
[44] JURIC D, SORIA J C, SHARMA S, et al. A phase 1b doseescalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors[J]. J Clin Oncol, 2014, 32(15_suppl): 9051.
[45]ALGAZI A P, ESTEVE-PUIG R, NOSRATI A, et al. Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma[J]. Pigment Cell Melanoma Res, 2018, 31(1): 110-114.
[46]SCHULER M, ZIMMER L, KIM K B, et al. Phase Ⅰb/Ⅱ trial of ribociclib in combination with binimetinib in patients with NRAS-mutant melanoma[J]. Clin Cancer Res, 2022, 28(14): 3002-3010.
[47]InxMed releases data demonstrating IN10018 therapeutic potential in patients with metastatic melanoma at SMR 2022[R]. SMR, October 17-20, 2022 (Abstract: 141).
[48]KINSEY C G, CAMOLOTTO S A, BOESPFLUG A M, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers[J]. Nat Med, 2019, 25(4): 620-627.
[49]RIBAS A, ALGAZI A, ASCIERTO P A, et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma[J]. Nat Commun, 2020, 11(1): 6262.
[50]GOGAS H, DRÉNO B, LARKIN J, et al. Cobimetinib plus atezolizumab in BRAFV600 wild-type melanoma: primary results from the randomized phase Ⅲ IMspire170 study[J]. Ann Oncol, 2021, 32(3): 384-394.
[51]GAO X J, XUE D D, CHENG J J, et al. Inhibition of axl promotes the therapeutic effect of targeted inhibition of the PI3K/Akt pathway in NRAS mutant melanoma cells[J]. J Oncol, 2022, 2022: 2946929.
[52]LI S, WU X, YAN X, et al. Toripalimab plus axitinib in patients with metastatic mucosal melanoma: 3-year survival update and biomarker analysis[J]. J Immunother Cancer, 2022, 10(2): e004036.
[53]PUZANOV I, SOSMAN J, SANTORO A, et al. Phase Ⅰ trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors[J]. Invest New Drugs, 2015, 33(1): 159-168.
[54]MAO L, GUO J, ZHU L, et al. A first-in-human, phase 1a dose-escalation study of the selective MEK1/2 inhibitor FCN-159 in patients with advanced NRAS-mutant melanoma[J]. Eur J Cancer, 2022, 175: 125-135.
[55]HAARBERG H E, PARAISO K H, WOOD E, et al. Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma[J]. Mol Cancer Ther, 2013, 12(6): 901-912.
[56]QIAN L, CHEN K, WANG C H, et al. Targeting NRAS-mutant cancers with the selective STK19 kinase inhibitor chelidonine[J]. Clin Cancer Res, 2020, 26(13): 3408-3419.

