BioRxiv:单氨基酸分辨率的长链蛋白质单分子纳米孔测序方法可检测完整折叠蛋白结构域
时间:2024-01-12 21:19:39 热度:37.1℃ 作者:网络
导读
蛋白质在细胞和生物体的生命活动过程中起着十分重要的作用,从生物的构成到生物的新陈代谢、遗传都和蛋白质的结构和功能密切相关。阐明蛋白质变异的复杂性对于理解生物学过程、识别疾病状态和开发有效的治疗方法非常关键。蛋白质多样性是一个术语,通常由于转录、翻译和翻译后修饰(PTM)的差异而存在的广泛蛋白质变异,这些变异可通过酶促和非酶促过程单独或结合地发生,进而在单个蛋白质分子上创建一个“PTM代码”,在推动生物过程中发挥独特作用。在天然、全长的状态下对单个蛋白质分子进行测序,将有助于人们更全面地了解蛋白质组多样性及其潜在编码机制,但目前的蛋白质测序技术还不能很好地实现这一目标。
近期,美国华盛顿大学的研究科研人员合作在BioRxiv发表了题为“Multi-pass, single-molecule nanopore reading of long protein strands with single-amino acid sensitivity”的文章。研究团队开发了一种新方法,有望利用Oxford Nanopore公司的商用平台实现完整蛋白质链的长距离(long-range)、单分子测序。具体而言,该方法使用ClpX去折叠酶(转位酶)使蛋白质通过CsgG纳米孔,实现了单氨基酸水平的敏感性,从而能够对长蛋白质链上的氨基酸替代组合进行测序。研究团队还开发了一种可多次重复读取相同的蛋白质链方法,以及一个生物物理模型,可基于氨基酸体积和电荷先验地模拟原始纳米孔信号,增强对原始信号数据的解读。综上,该方法可用于检测完整的折叠蛋白质结构域,进行完整的端到端分析。

文章发表在BioRxiv
文章作者华盛顿大学分子工程教授Jeff Nivala表示:“DNA和RNA纳米孔测序的重大突破是使用马达蛋白将DNA和RNA链拉过纳米孔。但对于蛋白质来说,这一步是一个挑战,因为蛋白质分子通常带有不均匀电荷,很难依靠电泳力以可控的方式将蛋白质链拉过孔。”
为克服读取全长蛋白质分子的挑战,研究团队使用Oxford Nanopore公司MinIon设备和R9.4流通池(flow cell)设计了一种两步方法(图1)。首先,通过电泳力使蛋白质底物从纳米孔中通过,同时在蛋白质C-末端固定一个阻断结构域,防止分子完全易位;随后,将ClpX(一种由ATP驱动的蛋白质去折叠酶)添加到阵列的顺式侧,以可控的方式将分析物从纳米孔中拉出(反式到顺式)。这种方法实现了长蛋白质链的连续易位,便于检测长达数百个氨基酸的蛋白质链中的多个单个氨基酸替代和PTM。
研究团队首先合成了一个蛋白质(P1)对该方法进行验证,发现该方法能够将捕获的蛋白质从C端向N端定向转运出纳米孔;在clpx介导的蛋白质通过纳米孔易位过程中,蛋白质尾部结构域的突变会引起离子电流状态的改变。研究团队进一步合成了三个新蛋白(P2、P3和P4),并使用动态时间扭曲(DTW)算法和t-SNE降维方法直接比较了上述四个蛋白质序列的信号谱。结果显示,易位信号之间的主要差异与不同蛋白质链上酪氨酸突变的位置相对应;每个独特的蛋白质序列产生的易位信号集不同。上述结果表明,使用这种方法可以观察到氨基酸序列依赖性信号特征。
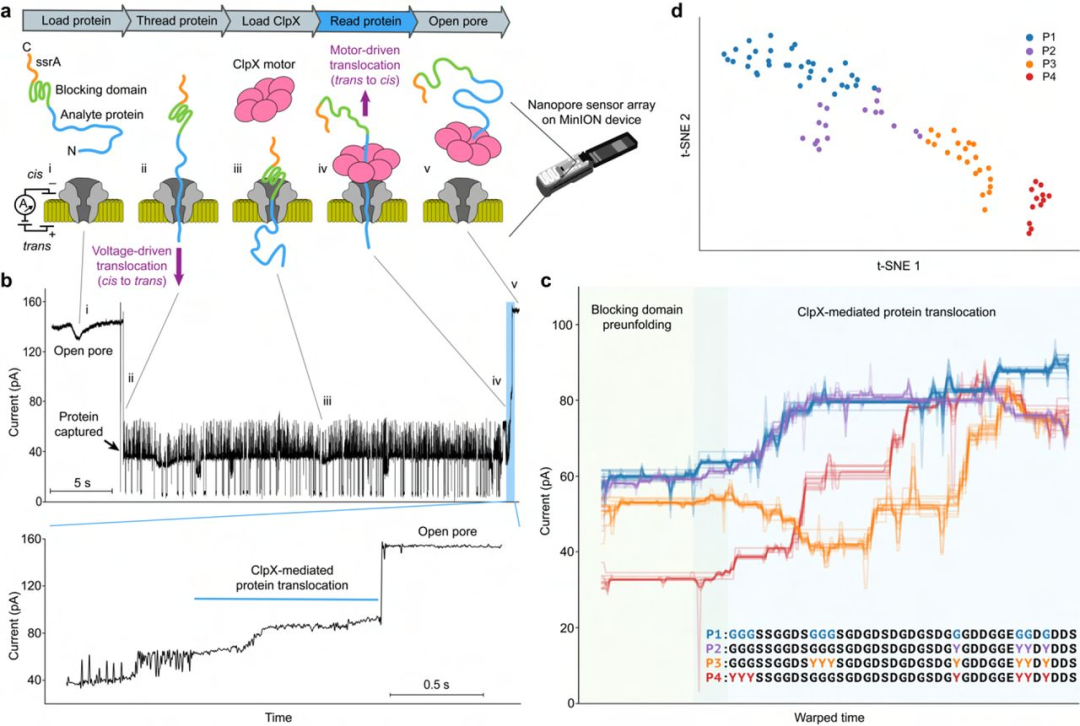
图1. 使用去折叠酶读取纳米孔蛋白质。
在建立了基于顺式的ClpX方法后,研究团队探究了该方法对单个氨基酸的敏感性,并为此设计了包含5个重复序列块的蛋白质构建体(PASTOR),每个重复序列块包含59个氨基酸,由甘氨酸、丝氨酸、天冬氨酸和谷氨酸的碱基序列构建;每个序列块的中心位置引入一个独特的氨基酸突变,并在每个末端使用双酪氨酸突变来划分序列块(图2)。 研究团队总合成8种不同的PASTOR变体(即HDKER、GNQST、FYWCP、AVLIM、VGDNY、TWAFH、PRMQE和KSILC),每种变体都包含不同的突变序列。
ClpX介导的PASTOR蛋白分析显示,由7个YY突变引起的重复模式出现延长的离子电流轨迹,在信号返回到开放通道之前可以观察到7个重复的dip;在这些dip之间,离子电流信号表现出独特且可重复的变化,且与每个序列块内的可变氨基酸突变相对应。利用YY突变的一致性,研究团队可将电流信号分为称为“YY dip”和“可变区域(VRs)”,进而能够单独分析每个氨基酸突变的独特离子特征。综上,该方法对单个氨基酸残基具有敏感性。
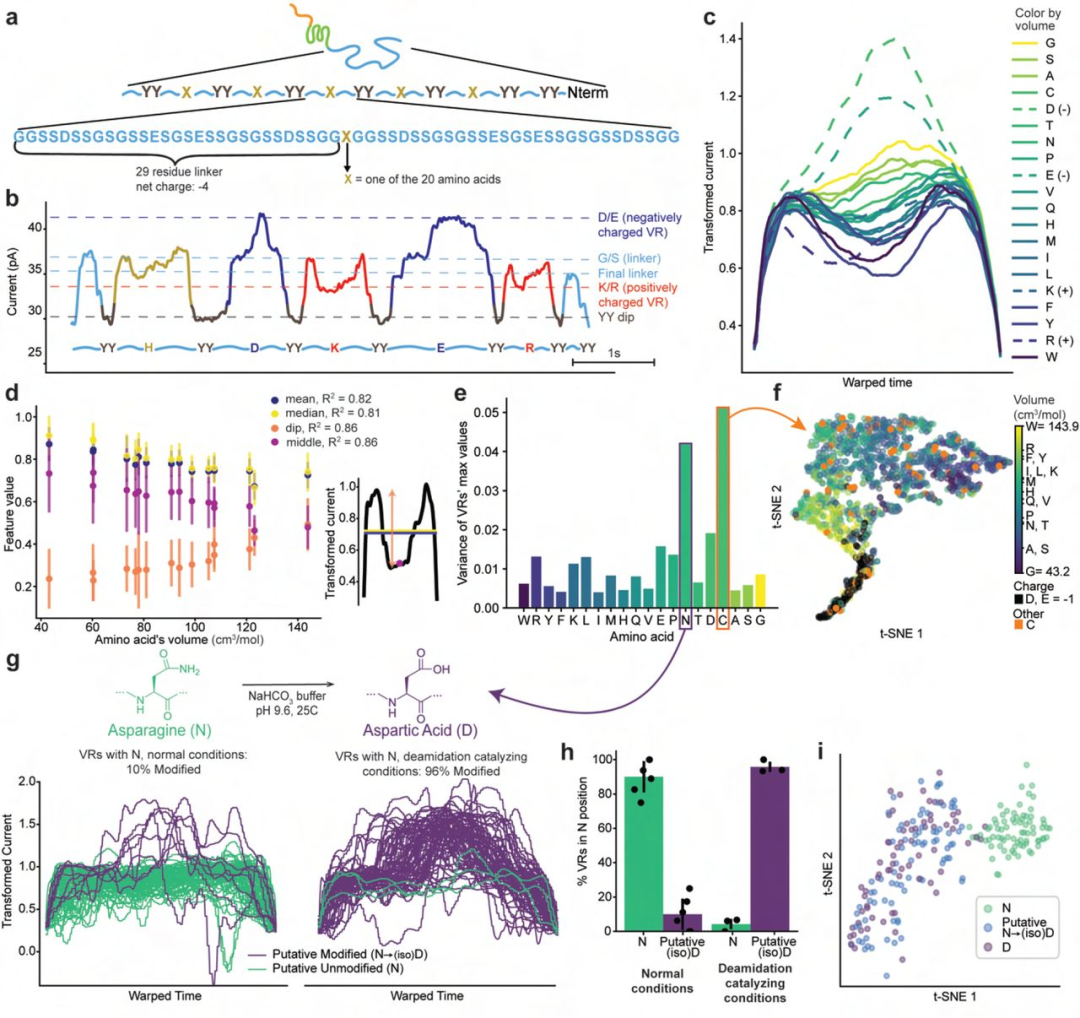
图2. 检测跨长蛋白链的单个氨基酸突变。
考虑到单个氨基酸体积与电荷之间的关系及其对纳米孔信号的影响,研究团队开发了一个生物物理模型,旨在直接模拟蛋白质氨基酸序列的纳米孔信号;该模型通过在一个移动的20个残基窗口内计算氨基酸体积和电荷的总和,并应用一个中心位置的负抛物线权重来确定信号(图3)。为定量评估生物物理模型与实际离子电流轨迹之间的一致性,研究团队计算了其在DTW对齐后的平均距离,并对模拟和实际电流轨迹进行了归一化处理。图3e是该模型为PASTOR-TWAFH蛋白序列生成的信号,与实际纳米孔轨迹对齐,这表明该模型能够在不同序列背景中准确模拟电流轨迹。
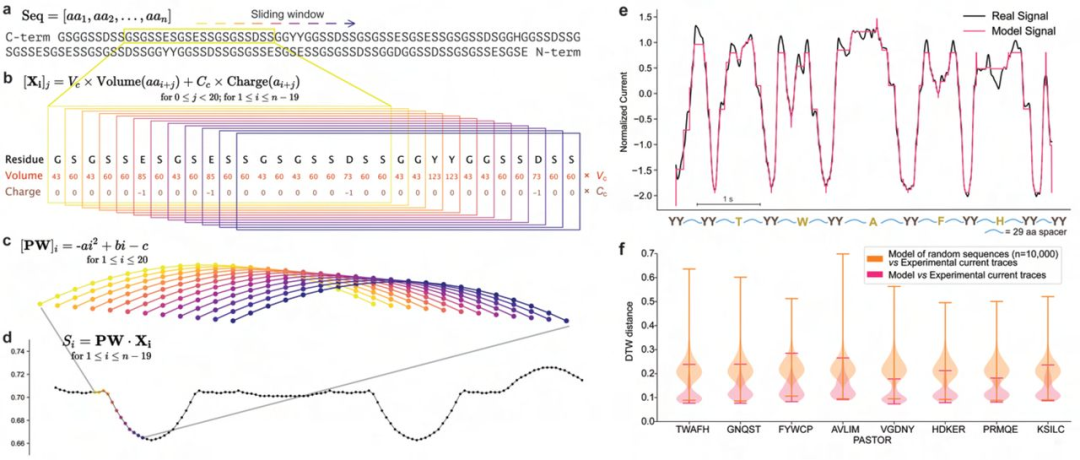
图3. 直接从蛋白质序列中模拟纳米孔离子电流轨迹的生物物理模型。
对PASTOR VRs进行测序是纳米孔蛋白测序发展的重要一步。为解决这一问题,研究团队首先通过训练机器学习模型来识别VRs中存在的单个突变,这一过程包括对原始信号迹进行过滤和缩放,对VR信号区域进行分割;进一步探索了几个经典的深度机器学习算法,发现随机森林模型的准确率最高(图4)。研究团队开始训练模型,对特定的5种氨基酸进性分类(例如G、V、W、R和D),模型准确率达到86%;随后将其扩大到20种氨基酸类型,表现最佳的模型性能显著优于虚拟分类器,准确率约28%,而后者的准确率为5.5%。此外,在20种氨基酸中,该模型的top-5分类准确率为67%,top-8分类准确率达到81%。
基于上述结果,研究团队将分类模型集成到PASTOR片段下游,开发出了一个端到端的 PASTOR "aminocaller",并对分类测试集中的一组PASTOR reads进行氨基调用。结果显示,HDKER序列中每个read的总体测序准确率平均约为62%(使用5种分类模型)和42%(使用20种分类模型),AVLIM序列中约为51%(使用5种分类模型)和21%(使用20种分类模型)。这些结果展示了纳米孔蛋白质读取的能力,不仅可以区分有限的一组氨基酸,还能在合成序列背景中对蛋白质链中的单个氨基酸进行测序。
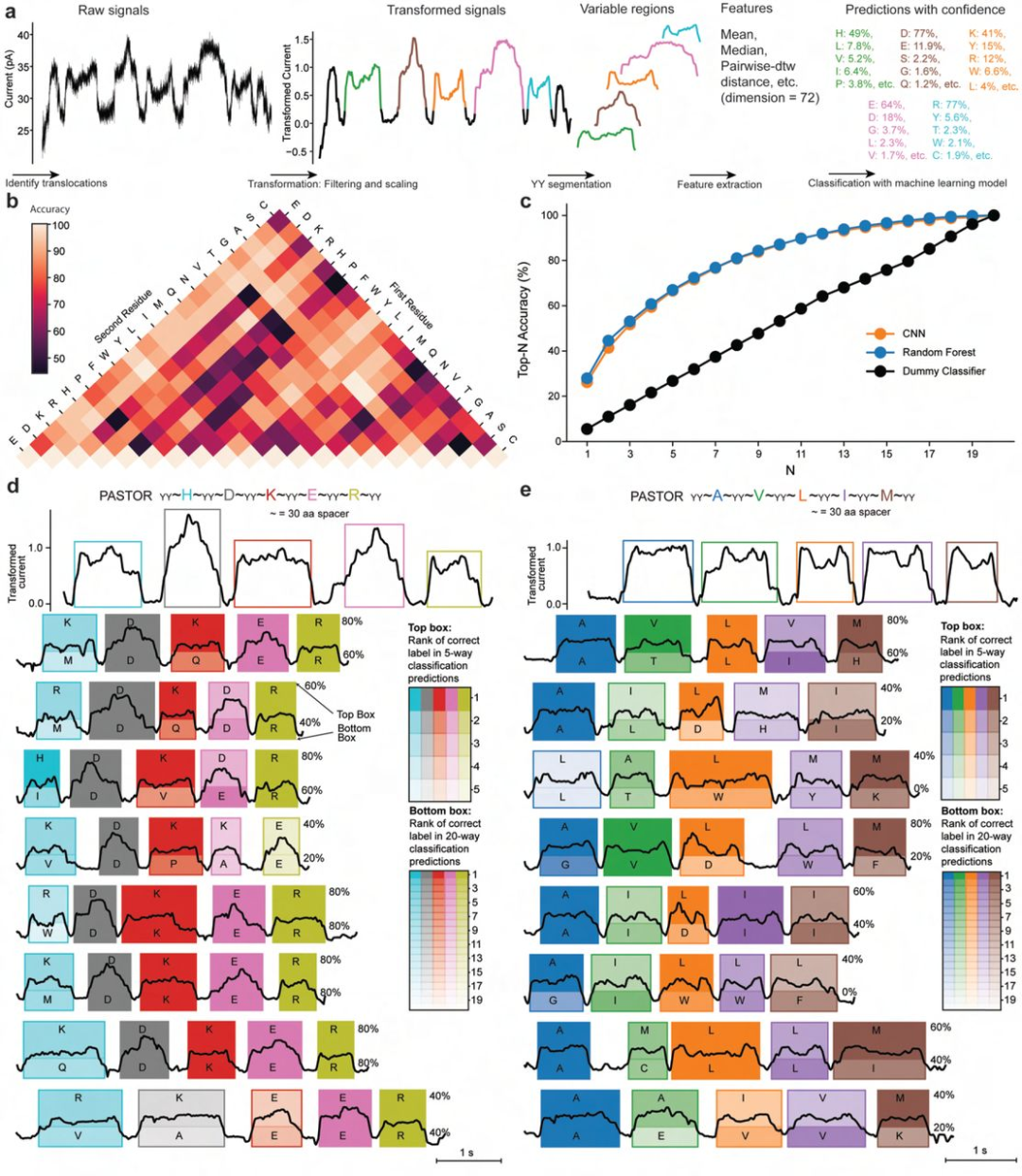
图4. 单个氨基酸突变的单分子纳米孔测序。
多读策略有助于在单个分子水平上生成一致的测序数据。因此,在建立了aminocaller后,研究团队开发了一种重读单个蛋白质分子的方法来提高单分子测序方法的准确性(图5):在PASTOR的N-末端附近添加了一个“滑动”的氨基酸序列(EPPPP),让蛋白质分子能够暂时滑离ClpX蛋白质;这使得底物蛋白可以通过电泳力再次穿回孔道,然后通过ClpX介导的逆易位进行重复测序读取。一旦ClpX重新抓住底物,重穿将停止,酶介导的易位将恢复。
最后,研究团队评估了单分子重读在提高测序准确性方面的潜力。结果显示,重新读取10次后,所有20种氨基酸分类任务的准确性从28%提高到61%(与5%的随机基线相比),这表明该重读方法能够提高单分子测序准确性。
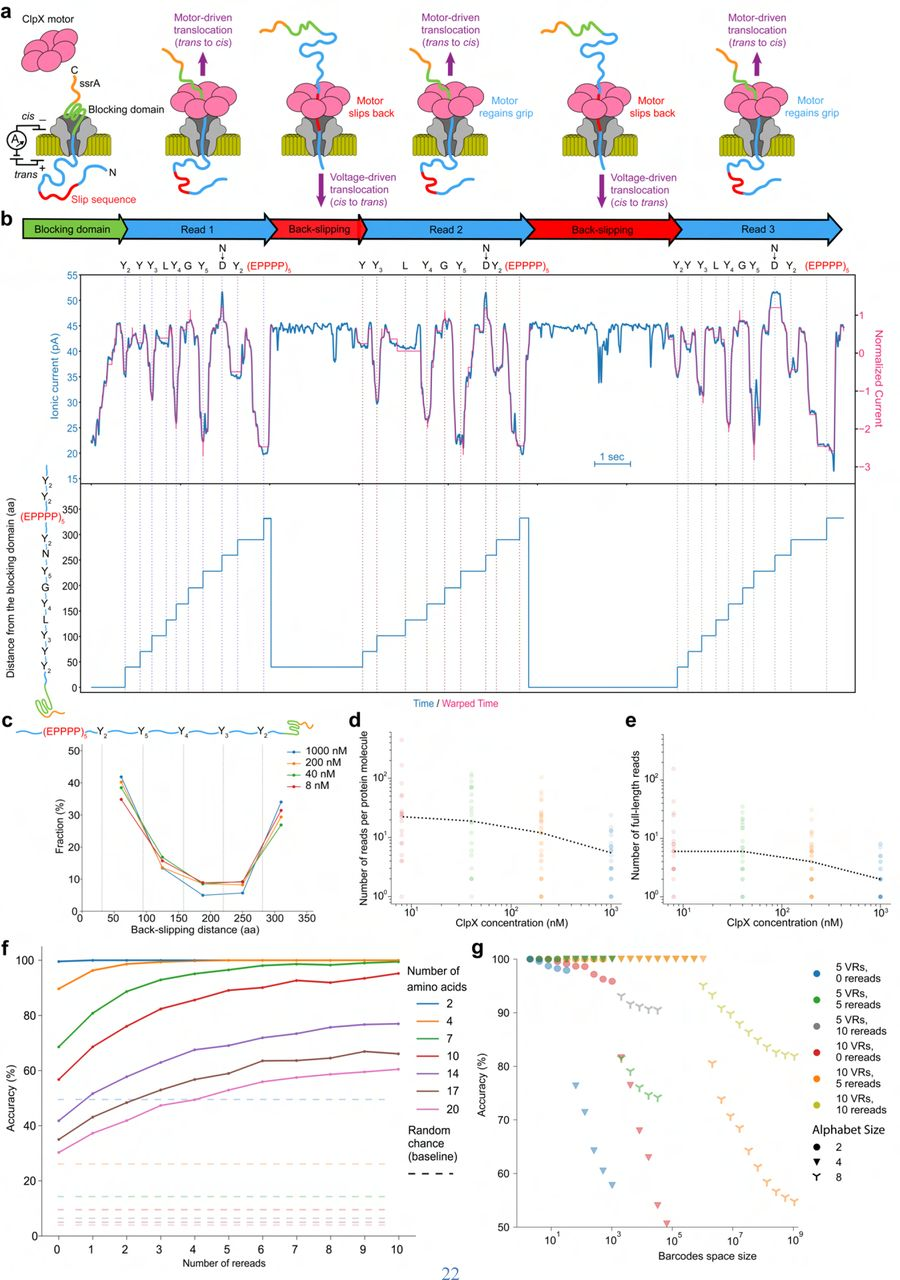
图5. 多次重读单个蛋白质分子。
综上所述,该研究介绍了一种使用纳米孔和去折叠酶马达蛋白,对长蛋白质链进行单分子读取的新方法。这种方法实现了对单个氨基酸的敏感性,并展示了在长蛋白质链上测序氨基酸替代的能力;有望为蛋白质条形码技术的进展开辟新的机会,并能以高精度进行解码。总之,该研究为全长蛋白质的鉴定奠定了基础,能够实现最高水平的蛋白质形态分辨率。
参考文献:
1.Motone, Keisuke et al. “Multi-pass, single-molecule nanopore reading of long protein strands with single-amino acid sensitivity.” bioRxiv : the preprint server for biology 2023.10.19.563182. 20 Oct. 2023, doi:10.1101/2023.10.19.563182. Preprint.
2.https://www.genomeweb.com/proteomics-protein-research/new-method-enables-long-range-protein-sequencing-using-oxford-nanopore

