高度可治的痴呆模拟病:LGI1抗体脑炎
时间:2024-08-04 06:01:16 热度:37.1℃ 作者:网络
论坛导读:AE是一种由免疫介导的以CNS症状为主要特征的自身免疫性疾病,常伴抗自身神经细胞内抗体和神经元表面抗体产生。前者为经典副肿瘤综合征抗体,包括抗Hu、Ma2、CV2、胶质纤维酸性蛋白(GFAP)及两性蛋白抗体,常伴随小细胞肺癌、乳腺癌以及淋巴瘤等,一般预后较差,抗肿瘤治疗有效,但对免疫治疗不敏感。而后者为新型AE,其抗体同时兼具致病性和特异性诊断标志物的性质,代表性自身免疫性抗体为抗‑N‑甲基‑D‑天冬氨酸受体(NMDAR)抗体、富亮氨酸胶质瘤失活蛋白1(LGI1)抗体、γ‑氨基丁酸B型(GABAB)受体抗体以及α‑氨基‑3‑羟基‑5‑甲基‑4‑异唑丙酸(AMPA)受体抗体等,伴或不伴肿瘤性疾病的发生,病程可逆,免疫治疗有效。
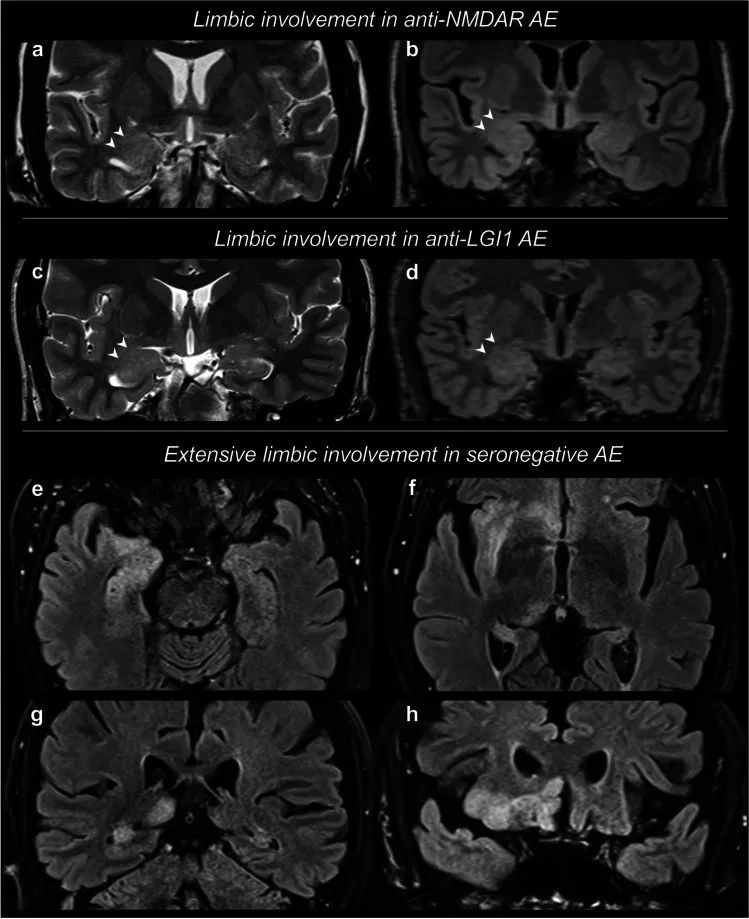
自身免疫性脑炎(Autoimmune encephalitis,AE)包括一组非感染性免疫介导的脑实质炎症性疾病,通常涉及皮质或深灰质,伴或不累及白质、脑膜或脊髓。其中是目前临床上最常见的AE脑炎,主要影响儿童和青年女性患者。针对GluN2B则导致Rasmussen脑炎。抗NMDAR脑炎与肿瘤相关,尤其是畸胎瘤,其次是卵巢外畸胎瘤和其他肿瘤。多数患者经免疫治疗后预后较好。
抗LGI1抗体脑炎是目前第二常见的AE脑炎。其主要症状包括记忆障碍、癫痫发作、精神和行为异常,常伴有低钠血症;面‑臂肌张力障碍样发作在诊断上具有特异性。LGI1是一种分泌的神经元蛋白,其IgG4亚型具有主要的致病作用。一般血清检测的敏感度较脑脊液高。抗LGI1抗体脑炎是认知和精神表现的可治疗和潜在可逆的原因,可能类似于老年患者的认知下降、快速进行性痴呆和复杂精神病。与其他表现为认知缺陷的疾病相比,这种病因与所需的替代治疗途径直接相关。
抗LGI1抗体脑炎通常表现为边缘性脑炎,主要表现为记忆和行为障碍,伴有睡眠障碍、颞叶癫痫发作,以及近30%的患者出现特征性面臂张力障碍性癫痫发作(FBDS)。尽管大多数患者经历了一个单相过程,随后恢复缓慢,经常(主要是认知)出现后遗症,但据报道约有15%-25%的患者出现复发。在临床实践中,辨别LGI1-Ab脑炎的残留症状和复发可能具有挑战性。复发的早期识别和治疗至关重要,因为它们可能导致额外的残疾和恶化预后。更重要的是,确定复发的风险因素将允许在疾病过程的早期实施预防策略,以完全避免复发的发生。
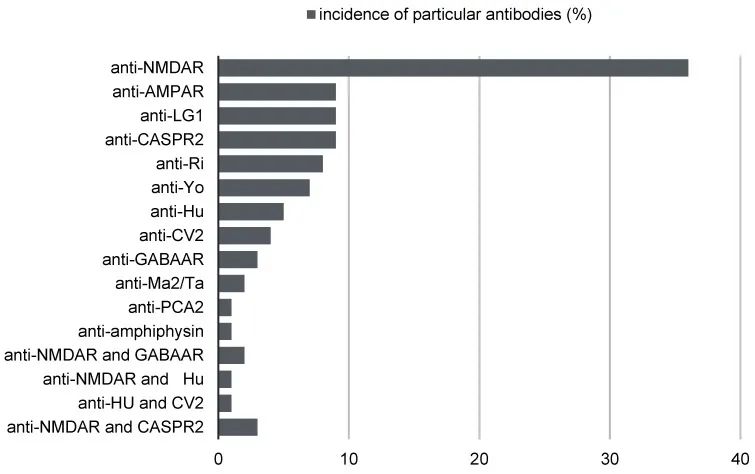
边缘性脑炎是一种病因不同的疾病,因为它既可以作为副肿瘤疾病发生,也可以发生在没有肿瘤疾病的患者中。在这种类型的脑炎患者中发现了各种神经元自身抗体。根据以前的研究,针对VGKC复合物的抗体是最常见的,并且在该复合物中包含的蛋白质中,针对LGI1蛋白质的抗体,LGI1蛋白质是一种通过参与突触前和突触后蛋白质复合物的形成而在突触传递中起重要作用的蛋白质。
近年来,自身免疫性脑炎越来越被认为是癫痫尤其是耐药性癫痫的病因之一。这种疾病的原因是针对抗原的自身抗体,抗原是离子通道、受体和其他参与神经元传递和脑可塑性的突触蛋白,有时也参与细胞毒性过程。带有文章中所列抗体的自身免疫性脑炎可以以多种方式表现出来。出现许多不同的症状并不罕见,包括神经和精神症状。存在抗体的自身免疫性脑炎的临床表现因其亚型和个体因素而异。
抗LGI1抗体脑炎是40岁以上最常见的自身免疫性脑炎。它的发病年龄中位数是65岁左右,通常表现为健忘症、性格改变和局灶性癫痫发作。像NMDAR-Ab-E一样,它可以通过免疫靶向治疗进行高度修改,但未经治疗会留下相当于痴呆症的缺陷。这是一种混合顺行性和逆行性记忆丧失的抽动模式。这可能伴随着执行功能障碍,以及广泛的神经精神影响,包括焦虑、抑郁和情绪不稳定(病理性哭泣)。可能会出现睡眠障碍,包括失眠和做梦/快速动眼期(REM)障碍。LGI1-Ab-E队列研究提供了支持早期免疫疗法消除癫痫发作和降低发展为固定认知缺陷风险的证据。
单相病程在特发性AE中更常见,而在一些副肿瘤综合征中,尤其是副肿瘤性小脑变性中,可以看到进展性病程,在癌症治疗后趋于平稳。已知癌症患者或癌症风险增加的患者(吸烟者、老年人和无诱因快速消瘦的患者)更容易发生副肿瘤性AE,而有其他自身免疫疾病个人或家族史的患者发生特发性AE的风险增加。之前有病毒感染、发烧或病毒样前驱症状很常见。复发的病程可能是自身免疫病因,但与多发性硬化和系统性炎症疾病的典型复发缓解过程不同,AE复发是罕见的,通常是由于治疗不足或免疫治疗的快速中断。

抗CASPR2和抗LGI1脑炎是老年人中最常见的亚型,大多数脑脊液检查结果正常。因此,在大多数情况下,在具有抗NMDAR、抗AMPAR和抗DPPX抗体的脑炎患者中,测试将显示脑脊液中的炎症变化,支持自身免疫性疾病的诊断。不幸的是,对于大多数抗LGI1和抗CASPR2抗体的患者来说,情况并非如此。然而,在老年人中出现抗LGI1和抗CASPR2抗体的疑似自身免疫性脑炎的情况下,这些测试的结果不应导致放弃血清抗神经元抗体测试的决定。根据2021年的最新建议,当临床怀疑自身免疫性脑炎时,该程序从脑成像和脑脊液(CSF)分析开始。诊断学的关键要素之一是检测抗神经元抗体的存在,不仅在脑脊液中,而且最重要的是在血清中。尽管有抗N-甲基-d-天冬氨酸受体抗体的显著鞘内合成,但血清中抗N-甲基-d-天冬氨酸受体抗体的绝对水平通常高于脑脊液。
CASPR2和LGI1是电压门控钾通道VGKC (VGKC)相关蛋白复合物的一部分。抗胞外蛋白LGI1和Caspr2的自身抗体在2010年首次被描述。这极大地改变了对电压门控钾通道相关抗体临床相关性的看法。自身免疫性脑炎伴有针对灭活的富含亮氨酸神经胶质瘤1 (LGI1)的自身抗体,可被诊断为抗LGI1自身免疫性脑炎。反过来,针对接触素样蛋白2 (CASPR2)的抗体的诊断负责抗CASPR2脑炎。与CASPR2抗体相关的脑炎是一种罕见的自身免疫性疾病。主要临床表现被认为是抽搐、记忆力丧失、精神症状、头晕和睡眠障碍。这两种亚型中最常见的临床症状是癫痫发作、记忆缺陷、精神障碍和意识障碍。目前,所有这些抗体都被归类为VGKC复合物抗体,并且通常被认为具有相似的临床价值
详细的病史和检查是AE诊断的第一步,也是最重要的一步。AE的免疫反应通常导致急性或亚急性表现,持续时间不到3个月。慢性表现仅见于其中一些疾病,尤其是LGI1、接触相关蛋白样2(CASPR2)、二肽基-p肽样蛋白(DPPX)和谷氨酸脱羧酶65(GAD65)-抗体性脑炎,否则应怀疑为神经退行性疾病或其他病因。同样,超急性表现也不典型,在这些情况下应考虑血管病因。
过去几年的研究表明,这些抗体介导的免疫反应具有不同的临床相关性。LGI1和Caspr2抗体与不同但明确的神经系统综合征相关。两者都有共同和不同的临床特征,由针对不同蛋白的自身抗体介导,并与电压门控钾通道复合。LGI1抗体一般以血液样本判读,如果血液和脑脊液同时阳性,可确诊,临床目前认为血液样本滴度大于1∶32且具有较典型的临床特征可确诊,低于1∶32需结合临床。
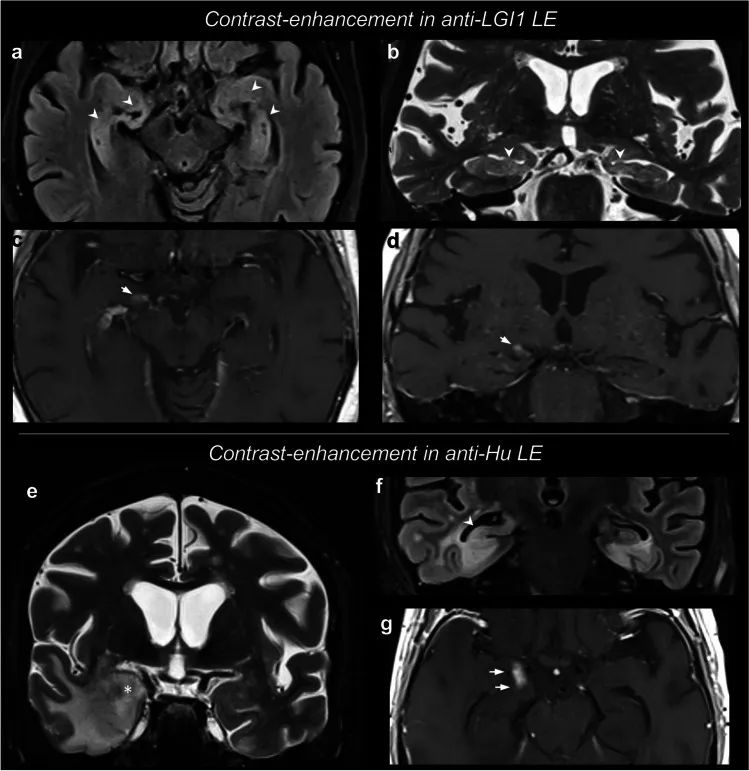
快速准确地诊断自身免疫性脑炎有助于迅速开始免疫治疗,以改善患者的预后。然而,仅临床特征可能不足以缩小鉴别诊断的范围,等待自身抗体结果可能会延误免疫治疗。一项横断面研究包括对来自英国牛津大学医院和美国梅奥诊所的192名患者的首个可用脑部磁共振成像(2000-2022年)的回顾性盲法分析。确定简单的磁共振成像(MRI)特征,准确区分两种常见形式的自身免疫性脑炎,LGI1-和caspr 2-抗体脑炎(LGI1/CASPR2-Ab-E ),与两种主要的鉴别诊断,病毒性脑炎(ve)和克雅病(CJD)。在这项研究中,T2和/或FLAIR高信号局限于颞叶,没有扩散限制或对比增强,有力地区分了LGI1/CASPR2-Ab-E和关键的鉴别诊断。这些观察将有助于加快免疫疗法的临床决策。它们对其他形式的自身免疫性脑炎和VE的普遍性应在未来的研究中进行检验。
在AE患者中,经验性静脉注射甲基强的松龙(剂量为1克/天,持续3-7天)是获得初始免疫抑制和抗炎效果的常见合理方法。对于已知对皮质类固醇有特异性反应的表现,即MRI上的脱髓鞘模式(提示AE与脱髓鞘综合征重叠),或斑点或放射状强化(分别提示CLIPPERS或自身免疫性GFAP星形细胞病),这也是首选方法。提示LGI1抗体脑炎的FBDS患者也可能对皮质类固醇有显著反应。
与经典肿瘤神经元抗体相关的已知或高度怀疑的副肿瘤性AE患者被认为主要是T细胞介导的炎症,理论上,皮质类固醇是免疫抑制的首选方案,而不是静脉注射IG或血浆置换(PLEX)。然而,与经典肿瘤神经元抗体相关的副肿瘤疾病通常对免疫抑制具有抵抗性,并且往往对癌症治疗反应最好。首次脑炎发作数年后可能复发,通常较轻,但会导致额外的残疾。皮质类固醇治疗降低了未来复发的风险,而初次发作后残留认知功能障碍的患者复发风险增加。
在过去的十年中,自身免疫性脑炎的诊断取得了重大进展。通过有效的鉴别诊断,获得了具有特定抗体亚型的自身免疫性脑炎的证据水平(可能的、很可能的或确定的),这可以导致快速免疫疗法的实施。早期诊断和早期开始靶向治疗增加了治疗过程成功的机会,并且可以帮助患者获得良好的预后。然而,为了改善自身免疫性脑炎的诊断和治疗,需要进一步的研究来开发更敏感的诊断工具,识别预后生物标志物并实施显著加速恢复的新治疗。
参考文献
Kelly MJ, etal. Magnetic Resonance Imaging Characteristics of LGI1-Antibody and CASPR2-Antibody Encephalitis. JAMA Neurol. 2024 May 1;81(5):525-533. doi: 10.1001/jamaneurol.2024.0126.
Abboud H,etal. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry. 2021 Jul;92(7):757-768. doi: 10.1136/jnnp-2020-325300.
Braczkowski M, etal. Autoimmune Encephalitis with Antibodies: Anti-NMDAR, Anti-AMPAR, Anti-GQ1b, Anti-DPPX, Anti-CASPR2, Anti-LGI1, Anti-RI, Anti-Yo, Anti-Hu, Anti-CV2 and Anti-GABAAR, in the Course of Psychoses, Neoplastic Diseases, and Paraneoplastic Syndromes. Diagnostics (Basel). 2023 Aug 3;13(15):2589. doi: 10.3390/diagnostics13152589.
中华医学会神经病学分会神经免疫学组.中枢神经系统自身免疫性疾病相关抗体检测专家共识2022.中华神经科杂志,2023,56(3):257-268.
Sanvito F, etal. Autoimmune encephalitis: what the radiologist needs to know. Neuroradiology. 2024 May;66(5):653-675. doi: 10.1007/s00234-024-03318-x.
Binks SNM, etal. LGI1-antibody encephalitis: how to approach this highly treatable dementia mimic in memory and mental health services. Br J Psychiatry. 2024 Jun;224(6):252-257. doi: 10.1192/bjp.2024.72.
Campetella L, etal. Predictors and Clinical Characteristics of Relapses in LGI1-Antibody Encephalitis. Neurol Neuroimmunol Neuroinflamm. 2024 May;11(3):e200228. doi: 10.1212/NXI.0000000000200228.

