罕见病专栏|青年男性,记忆力减退10个月余——成年脑型肾上腺脑白质营养不良
时间:2025-02-27 12:36:55 热度:37.1℃ 作者:网络
摘 要 本文报告了1例病程进展迅速的成人脑型肾上腺脑白质营养不良(adrenoleukodystrophy,ALD),患者表现为快速进展的记忆力下降,无其他阳性症状及体征,头颅磁共振见白质多发异常信号。因脑脊液白蛋白轻度升高,抗谷氨酸受体(NMDAR型)抗体阳性被误诊为 “自身免疫性脑炎”。2个月后复查磁共振见白质病灶范围增大,且部分强化。通过基因检测发现ABCD1基因c.1817C>T半合子变异被诊断为ALD,口服氢化可的松治疗,患者认知功能持续恶化,渐出现视力、听力损害,吞咽困难,二便失禁,发病后的26个月去世。通过对该病例的报道,提高医师对成人脑型ALD的认识,当男性患者出现快速进展的记忆力下降伴有白质脱髓鞘改变时,无论有无肾上腺皮质功能的异常,均应早期进行确诊性ABCD1基因检测。
关键词
肾上腺脑白质营养不良;肾上腺功能不全;ABCD1基因;自身免疫性脑炎;记忆力;脱髓鞘;认知障碍
1 临床资料
患者,男,29岁,因“记忆力减退10个月余”,于2018年6月28日就诊于上海市第六人民医院神经内科门诊。
2017年8月,患者被同事发现有记忆力下降,以近事记忆障碍为主,主要表现为日常工作需反复提醒。上述记忆力减退症状进行性发展,至2018年2月时明显加重,表现为经常反复询问同一件事情,在熟悉的地方迷路。2018年5月,患者至外院就诊,当时头颅MRI显示双侧侧脑室周围白质、桥脑背侧、双侧大脑脚、内囊后肢、胼胝体多发异常信号,考虑脑白质脱髓鞘病变。血皮质醇略低,ACTH正常。腰椎穿刺:脑脊液蛋白1 g/L(正常值:0.15~0.45 g/L),细胞数、氯化物和糖均正常。抗自身免疫性脑炎的抗体检测发现抗谷氨酸受体(NMDAR型)抗体阳性(1∶1)。外院诊断为“自身免疫性脑炎”,予甲强龙0.5 g/d及丙球20 g/d冲击治疗5 d后,患者出现肝功能损伤,认知障碍无改善。2018年7月因记忆力减退症状加重入我院。病程中,患者饮食睡眠可,大小便无异常,体重无明显变化。
既往史与家族史:出生史及既往史均无特殊,否认各类毒物及有害物质的接触史,家族内无类似病史。吸烟史4年,约20支/d,未戒烟,偶有少量饮酒。
体格检查:神志清晰,言语流利,构音清晰,对答切题,近记忆减退,时间定向减退,计算尚可。头发稍稀疏,双侧额纹、鼻唇沟对称,悬雍垂居中,双侧软腭上抬有力,咽反射存在,伸舌居中。四肢肌张力、肌力、感觉、深反射均正常,踝阵挛(-),共济运动正常。双侧病理征阴性,脑膜刺激征阴性。简易精神状态检查(MMSE)21分;蒙特利尔认知评估(MoCA)17分。
辅助检查:复查腰椎穿刺:蛋白511.6 mg/L;有核细胞数1.0×106/L(正常值:0~5×106/L);氯化物132 mmol/L(正常值:120~130 mmol/L);葡萄糖3.8 mmo/L(正常值:2.5~4.4 mmo/L)。8AM:皮质醇11.9 μg/dL(正常值:6.7~22.6 μg/dL);ACTH 218.99 pg/mL(正常值:7.0~65.0 pg/mL)。4PM:皮质醇4.87 μg/dL (正常值:2.9~17.3 μg/dL);ACTH 146.86 pg/mL(正常值:3.5~32.5 pg/mL)。入院后复查头颅MRI:双侧侧脑室周围白质、桥脑背侧、双侧大脑脚、内囊后肢、胼胝体多发异常信号,病灶范围较前有扩大,部分强化(图1)。脑电图及脑地形图正常。双侧肾上腺CT增强扫描:左侧肾上腺内侧肢略粗。脑组织立体定向活检显示,主要为大脑白质,水肿变性明显,可见较多泡沫样组织,个别血管周围淋巴细胞浸润。极长链脂肪酸(very long chain fatty acid ,VLCFA)检测,C26为5.71 nmol/mL(正常值:≤1.30 nmol/mL);C24/C22为1.89(正常值:≤1.39);C26/C22为0.13(正常值:≤0.023)。基因检查:高通量测序并经一代测序验证ABCD1基因c.1817C>T(p.Ser606Leu)半合子变异,Mutation taster预测此变异为有害突变,与肾上腺脑白质营养不良(adrenoleukodystrophy)相关。患者有一个姐姐和一个哥哥,哥哥32岁无症状,因缺少母亲和哥哥血样标本,只对其姐姐进行家系共分离验证,未发现携带该变异(图2)。
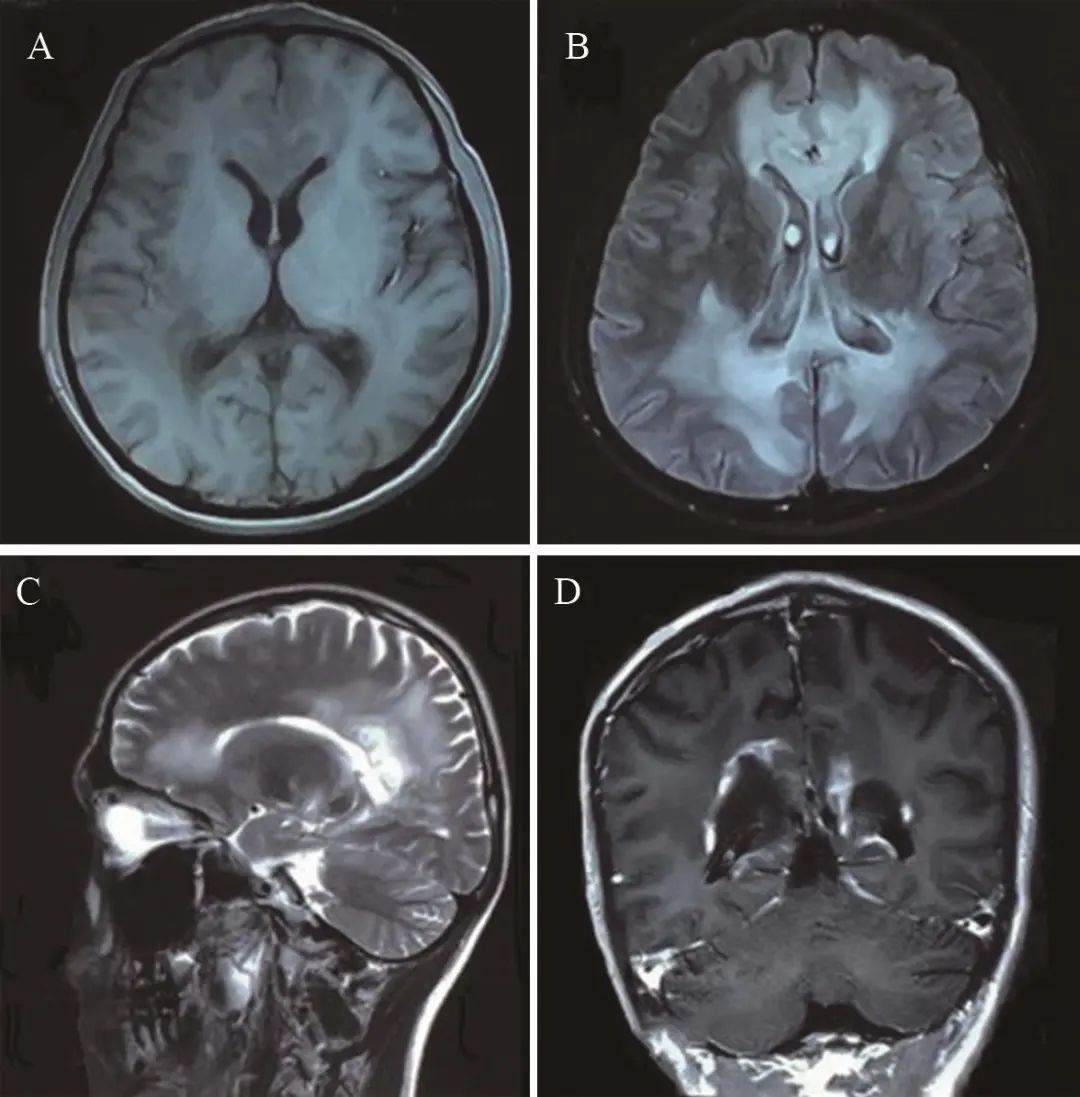
图1 头颅磁共振 A.磁共振T1加权像脑室周围、内囊后肢对称分布的低信号;B.磁共振Flair像脑室周围对称分布的蝶翼样高信号影;C.磁共振矢状位T2加权像胼胝体多发异常高信号影;D.磁共振T1增强扫描脑室周围部分病灶强化。
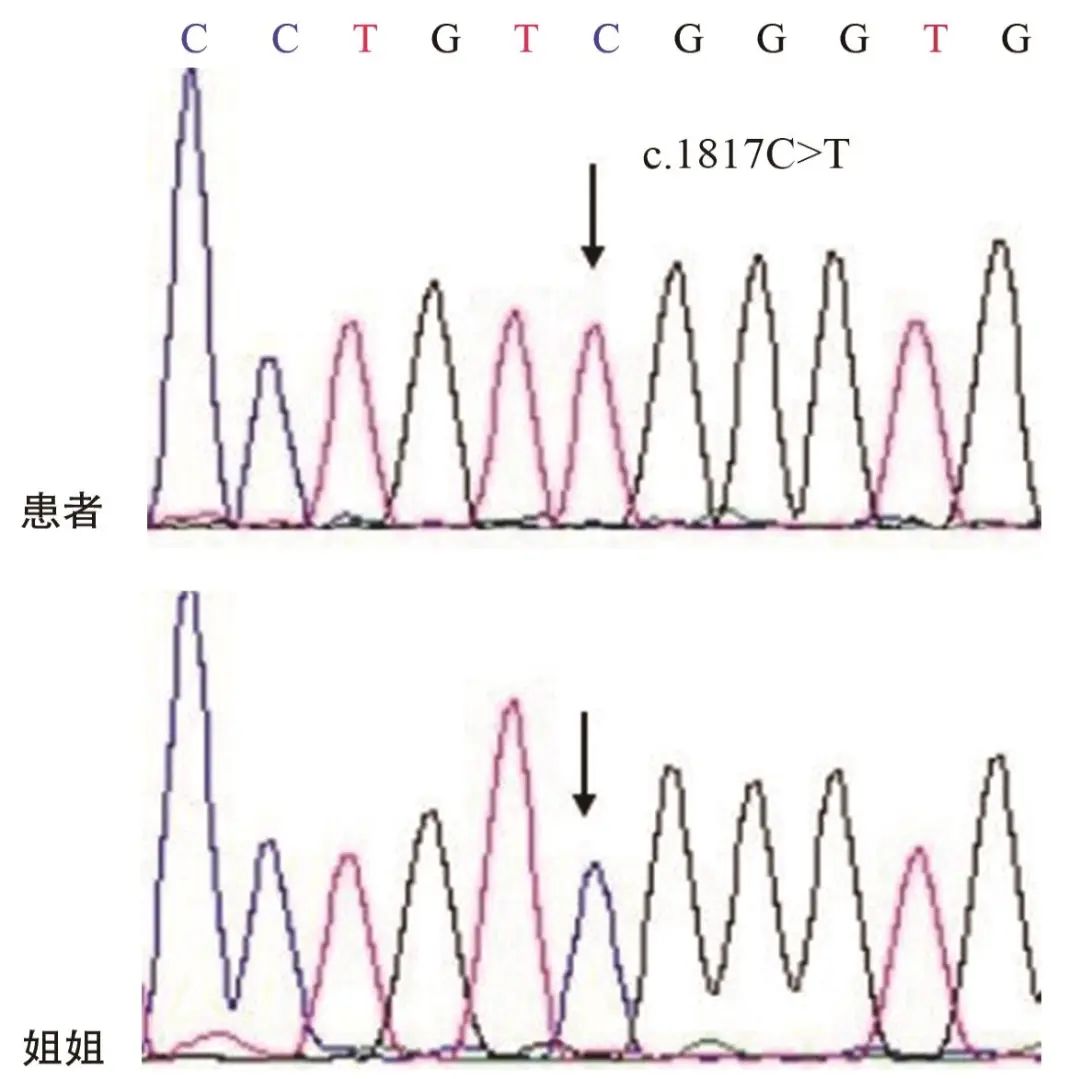
图2 基因测序图 患者ABCD1基因上存在一个半合子突变:c.1817C>T,其姐姐为野生型。
诊断肾上腺脑白质营养不良明确后,给与口服氢化可的松及饮食中控制极长链脂肪酸的摄入等对症支持治疗。患者出院后认知功能持续恶化,至2019年1月出现视力、听力下降,并伴有头部不自主晃动,6月出现意识不清,吞咽困难,大小便失禁,10月去世。
表1 MRI严重程度评分(LOES等[18])

注:每个区域的正常为0分,单侧受累为0.5分,双侧受累或萎缩为1分。最高分是34分。
2 讨论
肾上腺脑白质营养不良(adrenoleukodystrophy,ALD)是一种常见的过氧化物酶体病,发病率约为1:21000,无明显种族差异性,主要累及脑白质、肾上腺皮质和睾丸[1]。其致病基因ABCD1(ATP-Binding Cassette sub-family D,Member l)位于Xq28,由10个外显子和9个内含子组成,编码肾上腺脑白质营养不良蛋白(adrenoleukodystrophy protein,ALDP),该蛋白位于过氧化物酶体膜上,参与超长链脂肪酸(VLCFA;≥C22)的跨膜运输,其功能障碍会导致氧化功能受损,引起饱和VLCFA在细胞内积聚,特别是二十六烷酸(C26∶0)。
本病从儿童至成年均可发病,按照发病年龄及形式,可分为儿童脑型、肾上腺脊髓神经病型(adrenomyeloneuropathy,AMN)、成人脑型、单纯 addison病型、无症状女性携带者[2]。成年脑型ALD是一种急性炎症性脱髓鞘疾病,约占2%~5%,发病年龄多大于21岁,相较于儿童及青少年脑型的快速进展,其发病相对缓慢,早期很难发现认知功能的下降,随着疾病的进展,逐渐出现精神、认知及行为改变[3],平均在7.5年达到植物人状态和死亡[4-5]。该患者早期症状隐匿,出现记忆力下降症状后,病情快速进展,在仅仅2个月的时间内出现了认知功能障碍及影像学进展,在发病26个月时去世,其病情发展明显快于典型的成人脑型ALD的病程。患者ABCD1基因的8号外显子处发生了c.1817C>T错义突变,在既往的报道中,该突变既可见于快速进展的儿童脑型ALD,也可见于缓慢进展的AMN,即使在携带该突变的同一家系中,其表型也不一致[6],体现了该疾病的基因型-表型异质性。
患者血浆VLCFA水平升高,但C26∶0/C22∶0和C24∶0/C22∶0比率与脑病和肾上腺功能不全之间无明确关系[7],肾上腺皮质功能不全的终生患病率约80%[8]。该例患者在外院检查时皮质醇略低,而ACTH正常,结合患者的脑脊液自身免疫性脑炎抗体阳性,故认为患者不符合肾上腺脑白质营养不良的诊断。仅仅2个月后复查ACTH升高。肾上腺功能丧失是渐进的,最初多表现为ACTH升高。对ALD病情进展认识的不足是其误诊的原因。此外,患者脑脊液抗NMDAR IgG抗体(1∶1),其滴度很低,根据2017年提出《中国自身免疫性脑炎诊治专家共识》,该浓度为诊断自身免疫性脑炎的起始浓度,且患者除了认知功能障碍外,并无癫痫、肌张力障碍等自身免疫性脑炎的常见其他表现,在经过免疫治疗后,患者认知障碍无改善,基本可以排除自身免疫性脑炎的诊断。
ALD典型的磁共振表现为双侧脑室三角区周围对称性分布的长T1、T2信号影,累及胼胝体压部,双侧病灶相连,部分呈蝴蝶翼样[9],病灶常起源于顶枕区,渐向前累及颞叶、基底节及额叶,向下扩延累及脑干[10]。MRI T1对比增强与疾病进展呈强相关[11]。该例患者早期出现认知障碍时,磁共振表现为白质大片脱髓鞘,2个月后复查头颅MRI出现了病变范围的扩大,且部分病灶存在强化,这提示了疾病处于快速进展和活动期。神经炎症可能源于环境、遗传或表观遗传因素,导致巨噬细胞浸润并产生促炎细胞因子和趋化因子介质[12],使过氧化物酶体功能丧失,导致VLCFA的更多积累,VLCFA导致快速炎症性脑疾病的确切机制仍不清楚。本例患者脑组织定向立体活检脑白质水肿变性和血管周围淋巴细胞浸润,表明患者颅内脱髓鞘仍在进展,这也是患者在外院首次检查脑脊液时蛋白含量升高的原因。
脑型ALD是严重的高致死率疾病,未经治疗的患者5年存活率不到5%[13],目前尚无有效的治疗方案,在疾病早期进行同种异体造血干细胞移植(allogeneic hematopoietic stem cell transplantation,HSCT)能有效阻止脑型ALD的进展,但这不会逆转或修复已存在的颅内病变[14]。在神经症状出现前,约80%患者肾上腺皮质功能异常[15],根据肾上腺脑白质营养不良患者肾上腺功能不全的临床实践指南[16],当基线ACTH>300 pg/mL,且皮质醇<18 μg/dL时,需皮质类固醇替代治疗。口服洛伦佐油,即三油酸甘油酯和三酸甘油酯的4∶1混合物,并结合饮食中限制VLCFA的摄入时,可以使患者血浆VLCFA水平正常化[17]。目前关于洛伦佐油疗效仍存在争议,经过尸检发现,洛伦佐油对脑内VLCFA浓度的影响远小于血清,故多数的观点认为,不应把洛伦佐油作为治疗和预防ALD病程进展的常规治疗。
3 点评
成年发病的脑型ALD是一种罕见的高致死性疾病,在疾病早期部分患者肾上腺皮质功能正常,临床医师对其认识不充分,易造成疾病误诊。本例患者提示我们,对有进行性认知障碍,并伴有颅内对称性脱髓鞘病变的男性患者,虽肾上腺功能正常,仍应警惕此病的可能性。当检测到VLCFA浓度升高时,应进行确诊性ABCD1基因检测。此病目前尚无确切的治疗方法,在疾病早期进行HSCT是目前可能挽救患者生命的唯一方法,这就要求我们临床医生,提高对此病的认识,早期诊断,早期干预。
参考文献:
1. BEZMAN L, MOSER A B, RAYMOND G V, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening[J]. Ann Neurol, 2001, 49(4): 512-517.
2. KÖHLER W, CURIEL J, VANDERVER A. Adulthood leukodystrophies[J]. Nat Rev Neurol, 2018 , 14(2): 94-105.
3. 尤桦菁, 李洵桦. 成年男性行走不稳、记忆力下降及视物障碍半年——X连锁肾上腺脑白质营养不良[J]. 中国神经精神疾病杂志, 2020, 46(5): 318-320.
4. TAKEMOTO Y, SUZUKI Y, TAMAKOSHI A, et al. Epidemiology of X-linked adrenoleukodystrophy in Japan[J]. J Hum Genet, 2002 , 47(11): 590-593.
5. ENGELEN M, KEMP S, DE VISSER M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management[J]. Orphanet J Rare Dis, 2012 , 7: 51.
6. 平莉莉,包新华,王爱花,等. X连锁肾上腺脑白质营养不良89例临床特征及基因型/表型关系[J]. 中华儿科杂志, 2007, 45(3): 203-206.
7. TRAN C, PATEL J, STACY H, et al. Long-term outcome of patients with X-linked adrenoleukodystrophy: A retrospective cohort study[J]. Eur J Paediatr Neurol, 2017, 21(4): 600-609.
8. HUFFNAGEL I C, LAHEJI F K, AZIZ-BOSE R, et al. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration[J]. J Clin Endocrinol Metab, 2019 , 104(1): 118-126.
9. 江文静, 迟兆富, 杜滨锋, 等. 磁共振成像在肾上腺脑白质营养不良诊断中的应用 [J]. 中华神经科杂志, 2008, (2): 106-109.
10. GARSIDE S, ROSEBUSH P I, LEVINSON A J, et al. Late-onset adrenoleukodystrophy associated with long-standing psychiatric symptoms [J]. J Clin Psychiatry, 1999, 60(7): 460-468.
11. MELHEM E R, LOES D J, GEORGIADES C S, et al. X-linked adrenoleukodystrophy: the role of contrast-enhanced MR imaging in predicting disease progression [J]. AJNR Am J Neuroradiol, 2000, 21(5): 839-844.
12. SINGH I, PUJOL A. Pathomechanisms underlying X-adrenoleukodystrophy: a three-hit hypothesis [J]. Brain Pathol, 2010, 20(4): 838-844.
13. MOSER H W. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy[J]. Brain, 1997, 120 (Pt 8): 1485-1508.
14. KEMP S, HUFFNAGEL I C, LINTHORST G E, et al. Adrenoleukodystrophy - neuroendocrine pathogenesis and redefinition of natural history[J]. Nat Rev Endocrinol, 2016, 12(10): 606-615.
15. DUBEY P, RAYMOND G V, MOSER A B, et al. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening [J]. J Pediatr, 2005, 146(4): 528-532.
16. REGELMANN M O, KAMBOJ M K, MILLER B S, et al. Adrenoleukodystrophy: Guidance for Adrenal Surveillance in Males Identified by Newborn Screen[J]. J Clin Endocrinol Metab, 2018, 103(11): 4324-4331.
17. MOSER H W, BOREL J. Dietary management of X-linked adrenoleukodystrophy[J]. Annu Rev Nutr, 1995, 15: 379-397.
18. LOES D J, HITE S, MOSER H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations [J]. AJNR Am J Neuroradiol, 1994, 15(9): 1761-1766.
【引用格式】刘桃桃 ,刘晓黎,曹立. 青年男性,记忆力减退10个月余——成年脑型肾上腺脑白质营养不良[J]. 中国神经精神疾病杂志,2022,48(2):125-128.
【cite this article】LIU T T ,LIU X L,CAO L. Young male with memory loss for more than 10 months——adult cerebral adrenoleukodystrophy[J]. Chin J Nervous Mental Dis, 2022, 48(2): 125-128.
DOI:10.3969/j.issn.1002-0152.2022.02.013

