Cancer Discov:于吉洋团队揭示肿瘤细胞GPR65失活驱动CAR-T治疗B型急淋白血病复发耐药机制
时间:2025-02-28 12:17:21 热度:37.1℃ 作者:网络
CAR-T细胞疗法在治疗血液肿瘤方面取得一定成果,但仍有部分患者复发或无响应。肿瘤异质性及肿瘤与微环境相互作用对CAR-T细胞反应的影响尚不清楚,需探究其复发耐药机制以改进治疗方法。
GPR65是一种质子感应细胞表面受体,在酸性刺激下可刺激环磷酸腺苷(cAMP)的形成。此前的一些研究表明,在部分实体肿瘤细胞系中GPR65被鉴定为肿瘤存活因子,然而在血液系统肿瘤中它却能够抑制肿瘤的生长和存活。
2025年2月25日, 美国圣裘德儿童研究医院 于吉洋 课题组 和 Terrence Geiger 课题组研究人员在 Cancer Discovery 上发表题为 GPR65 inactivation in tumor cells drives antigen-independent CAR-T cell resistance via macrophage remodeling 的研究论文,揭示了GPR65在CAR-T治疗耐药中起关键作用,其缺失会通过重塑肿瘤微环境中的巨噬细胞导致CAR-T细胞耐药,为B-ALL治疗提供了新的理论依据和潜在治疗靶点。

作者将两种不同来源的B-ALL肿瘤克隆(m.PR和m.CR)移植到小鼠体内,并用CAR-T细胞治疗。结果显示m.CR肿瘤对CAR-T细胞治疗完全响应,m.PR肿瘤虽初期有响应但最终复发。分析发现m.CR肿瘤在基线时高表达CD19,复发的m.PR肿瘤则丢失CD19。RNA-seq分析表明m.CR肿瘤中GPR65等GPCR信号相关基因显著上调,功能富集分析也显示GPCR相关通路在m.CR肿瘤中富集,且m.CR肿瘤中胞质cAMP和CREB S133磷酸化水平相对m.PR细胞增加。该部分探究了B-ALL中GPCR信号与CAR-T细胞治疗临床前反应的关系,发现GPR65可能是关键的响应驱动因子。

图1:B-ALL GPCR信号可预测CAR-T细胞治疗的临床前反应
为确定GPR65在人类肿瘤反应中的相关性,作者研究分析了接受CD19 CAR-T或blinatumomab治疗患者的临床试验数据,同时利用小鼠模型中m.PR和m.CR B-ALL克隆的相关数据,采用三步策略筛选CAR-T响应特征基因。主成分分析(PCA)可有效区分CAR-T治疗响应者和非响应者;整合分析将GPR65列为与两种物种响应者紧密相关的首要候选基因,且基因特征可准确分层CD19 CAR-T治疗患者,GPR65活性和表达在响应者中显著更高;ROC分析进一步证明其对患者CAR-T治疗反应的预测能力,并且在成人B-ALL患者对blinatumomab响应中也有类似表现。该部分通过系统信息学分析,探究B-ALL中GPCR信号与靶向CD19免疫治疗应答的关系,发现GPR65 可能是关键的响应驱动因子。
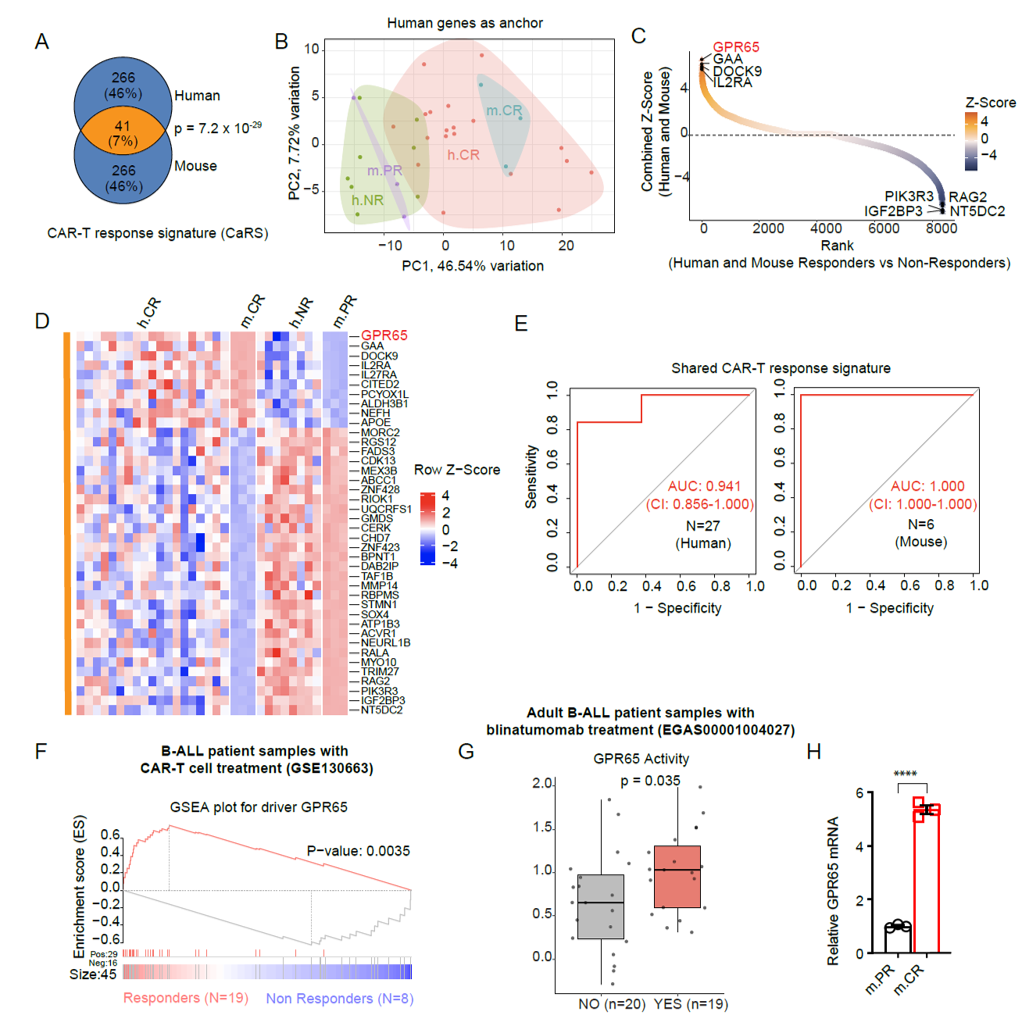
图2:GPR65是B-ALL中重要的CAR-T反应驱动因子
为了进一步研究GPR65基因在肿瘤应答中的作用,作者敲除m.CR hCD19+ B-ALL细胞系中GPR65基因。体内实验中,m.CR和GPR65 KO肿瘤的肿瘤负荷相似,但GPR65 KO肿瘤对CAR-T治疗无反应,而m.CR肿瘤可完全消退。相反,在GPR65 KO肿瘤中过表达GPR65可恢复其对治疗的敏感性。在CAR-T治疗4天后,对小鼠体内肿瘤和 CAR-T细胞数量进行检测,发现GPR65 KO肿瘤小鼠各器官的肿瘤负荷高于m.CR肿瘤小鼠且生存率更低。同时,GPR65 KO肿瘤小鼠的CAR-T细胞数量并不少于m.CR肿瘤小鼠,表明GPR65 KO肿瘤对治疗抵抗并非由于CAR-T细胞存活或扩增受抑制。该部分实验表明GPR65缺失的肿瘤细胞其抗性并非抗原下调或CAR-T细胞扩增被抑制。
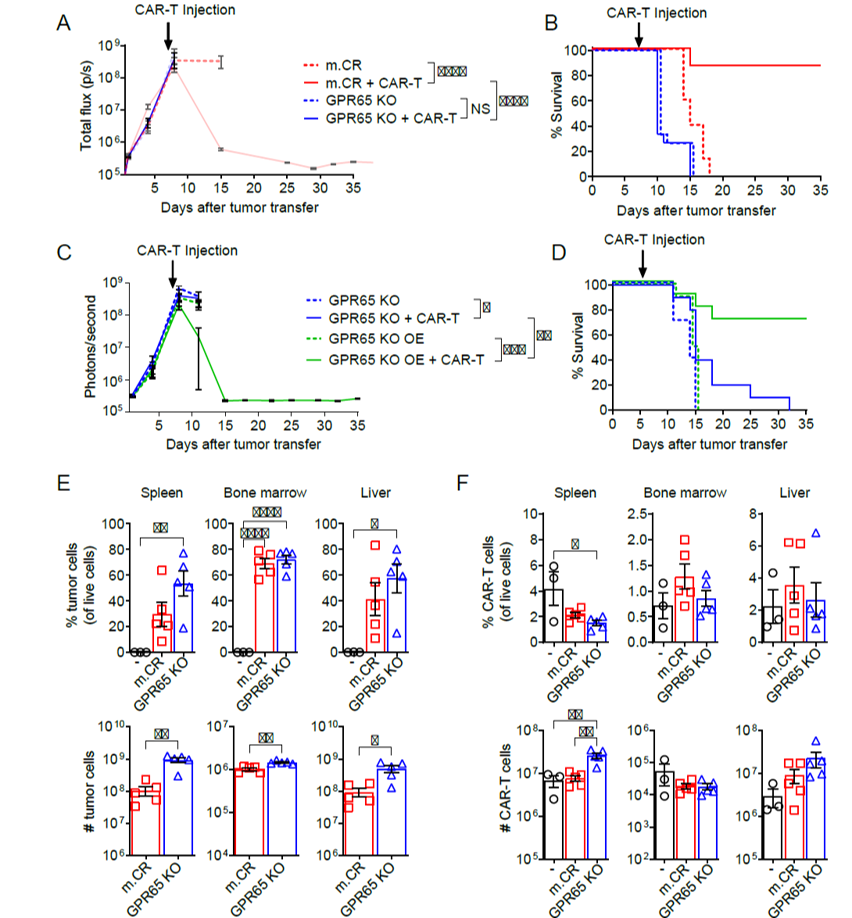
图3:GPR65敲除肿瘤驱动体内CAR-T抵抗而非减少抗原表达或抑制CAR-T扩增
在对GPR65 KO肿瘤小鼠的CAR-T细胞研究中发现,尽管这些细胞无法清除GPR65 KO肿瘤,但仍保留了效应功能,推测GPR65 KO肿瘤细胞并非通过下调CD19表达或损害CAR-T细胞的扩增和功能来产生抗性,而是可能通过改变肿瘤微环境介导了CAR-T细胞抗性。利用scRNA-seq对m.CR和GPR65 KO肿瘤及肿瘤微环境(TME)进行分析,揭示了两者在肿瘤细胞特性和TME细胞组成上的差异。
对宿主免疫细胞群体组成分析发现,在CAR-T治疗后,巨噬细胞群体在GPR65 KO肿瘤小鼠TME中的数量比m.CR肿瘤小鼠TME中增加了2.96倍,变化最为显著。进一步对巨噬细胞进行scRNA-seq分析,发现CAR-T治疗会诱导巨噬细胞群体扩增,GPR65 KO组巨噬细胞数量更多,且巨噬细胞存在异质性。来自m.CR肿瘤小鼠的巨噬细胞表达促炎型巨噬细胞(M1)相关基因,而GPR65 KO肿瘤小鼠的巨噬细胞表达抗炎型巨噬细胞(M2)相关基因。
通过GSEA分析发现 m.CR肿瘤巨噬细胞呈M1样表型,GPR65 KO肿瘤巨噬细胞呈M2样表型,在患者样本中也观察到类似现象,即CD19+复发患者骨髓巨噬细胞呈M2样表型,而CR患者呈M1样表型,表明巨噬细胞极化可能是肿瘤对CAR-T细胞治疗产生抗性的机制之一。
为确定巨噬细胞在GPR65 KO肿瘤小鼠体内对CAR-T细胞治疗的影响,在CAR-T治疗前用抗-CSF1R(CD115)抗体耗竭巨噬细胞。巨噬细胞耗竭后,原本对CAR-T细胞治疗无反应的GPR65 KO肿瘤小鼠和m.CR肿瘤小鼠一样,能够完全响应治疗且无肿瘤复发。这表明在GPR65 KO肿瘤微环境中,扩增且呈M2极化的巨噬细胞池是导致CAR-T细胞治疗抗性的必要条件,消除巨噬细胞可恢复CAR-T细胞的肿瘤杀伤活性。

图4:巨噬细胞极化是CAR-T治疗抵抗的机制之一
细胞间通讯分析表明,VEGFA在GPR65 KO肿瘤与宿主巨噬细胞和单核细胞的通讯相较于m.CR肿瘤有所增加。其中,VEGFA通讯和表达在GPR65 KO肿瘤中尤为突出。为确定 VEGFA在GPR65 KO肿瘤细胞治疗抗性中的作用,研究人员构建了m.CR和GPR65 KO细胞的VEGFA基因敲除细胞系,并将其移植到免疫健全小鼠体内,随后进行CAR-T细胞治疗。结果发现,m.CR和VEGFA KO肿瘤小鼠对CAR-T细胞治疗完全响应,而GPR65 KO肿瘤小鼠仍对CAR-T 治疗产生抵抗。尽管DKO肿瘤对CAR-T治疗反应不完全,在初始缓解之后仍会复发,但DKO肿瘤小鼠相较于GPR65 KO肿瘤小鼠生存期明显延长。且DKO肿瘤在治疗4天后的肿瘤细胞和巨噬细胞数量相较于GPR65 KO肿瘤有所减少。该部分的研究结果表明,GPR65 KO肿瘤细胞通过上调VEGFA促进CAR-T细胞治疗抗性。
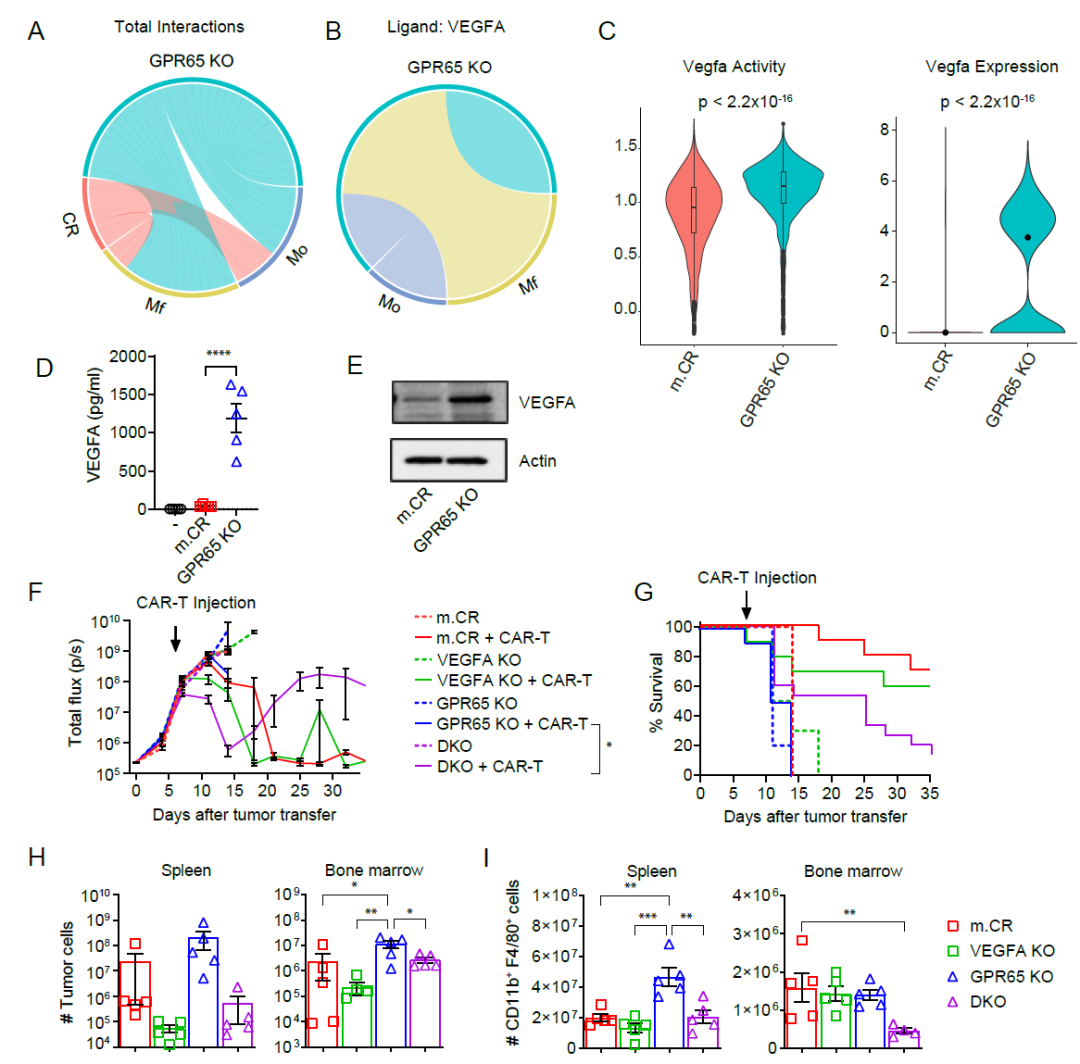
图5:GPR65 KO肿瘤细胞通过上调VEGFA促进CAR-T细胞治疗抗性
鉴于VEGFA缺失在DKO肿瘤中可使肿瘤对CAR-T细胞敏感,研究进一步探究抗VEGFA 药物是否能恢复GPR65 KO肿瘤对CAR-T细胞治疗的敏感性。结果显示,与接受同型对照的小鼠相比,联合使用CAR-T细胞和抗VEGFA抗体显著延长了GPR65 KO肿瘤小鼠的生存期,并降低了肿瘤负荷。对于m.CR肿瘤小鼠,抗VEGFA治疗不影响其对CAR-T细胞治疗的响应性。虽然GPR65 KO肿瘤小鼠在联合治疗后最终仍会因肿瘤进展而死亡,但这表明抗 VEGFA抗体联合CAR-T细胞是一种减轻GPR65相关CAR-T细胞治疗抗性的有效策略。
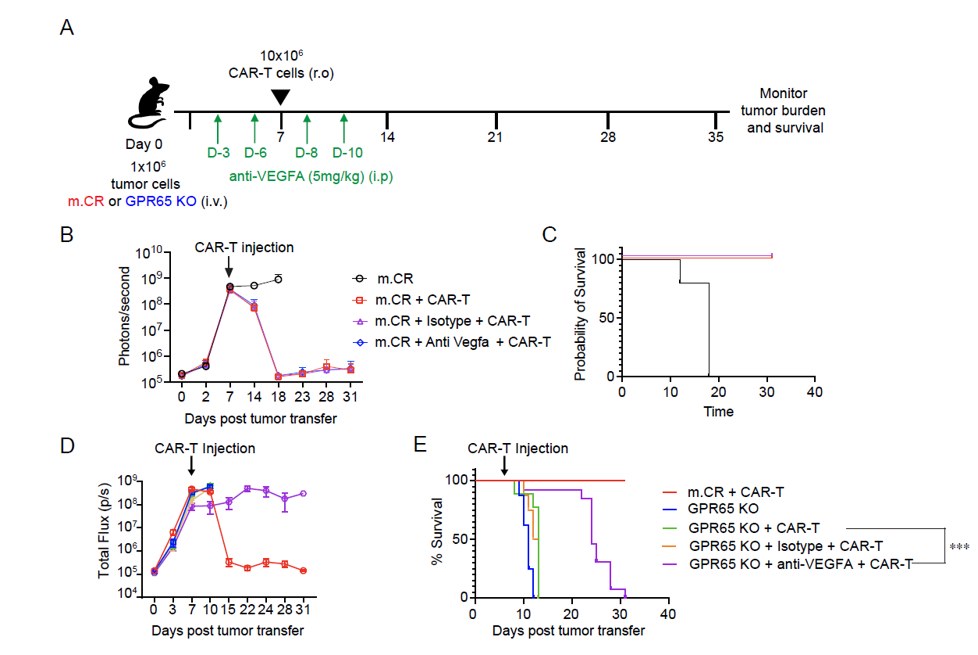
图6:抗VEGFA使GPR65 KO肿瘤对CAR-T细胞治疗增敏
综上,针对不分患者对CAR-T治疗无效或治疗后复发的问题,研究人员发现GPR65是B-ALL对CAR-T细胞治疗反应的关键因素,其缺失会通过改变肿瘤微环境(如巨噬细胞重塑和VEGFA上调)导致CAR-T细胞治疗抵抗。靶向VEGFA或巨噬细胞为优化CAR-T细胞治疗提供了新途径,并且GPR65有望成为预测患者治疗反应的生物标志物。

