妇瘤新视野丨解密Peutz-Jeghers综合征
时间:2023-04-21 10:27:48 热度:37.1℃ 作者:网络
一、病例简介
主诉:
62岁女性绝经后阴道出血伴下腹痛50余天。
既往史:
2型糖尿病;否认肿瘤家族史,否认家族性胃肠道息肉病史,否认胃肠道或者宫腔息肉手术史。
体格检查:
生命体征平稳,全身皮肤粘膜未见色素沉着病灶,心肺未及异常,腹软,无压痛及反跳痛。
妇科检查:
外阴已婚已产型,发育正常,未见色素沉着;阴道粘膜光滑,未见色素沉着;宫颈正常大小,光滑,触血阴性,穹窿萎缩变浅;子宫前位,饱满,活动欠佳,无压痛;盆腔偏左侧可及一肿物,上极达脐水平,不活动;盆腔偏右侧可及一囊性肿物,上极达髂前上棘水平,可活动,无压痛;肛查道格拉斯窝未及明显结节,直肠粘膜光滑。
辅助检查:
TCT:良性反应性改变;HPV:阴性。
CA125:10.96 U/ml;CA199:4.72 U/ml;CEA:1.95 ng/ml。
盆腔超声:
盆腔内多房囊性包块,性质待定;子宫内膜增厚,回声不均。
盆腔MRI:
子宫内膜薄厚不均,上段最厚0.8 cm;宫颈大小及形态正常,黏膜欠规整,DWI稍高信号;数个微小纳囊;子宫后上方及右上方各见囊性肿块,大小约9.2 cm*14.2 cm*7.7 cm、9.2 cm*5 cm*8 cm,囊内多发分隔,囊液大部分呈较长T1较长T2信号,部分呈等或短T1长T2信号;囊壁及分隔大部分不均匀较薄,左侧部分增厚,轻度至明显强化,肿块达盆壁双侧及右侧髂窝;双侧髂血管旁数个小淋巴结。
MRI影像提示子宫后上方及右上方多房囊性肿块,考虑双附件来源肿瘤,卵巢上皮性肿瘤可能大,右侧附件肿瘤偏良性、左侧交界性可能,黏液性肿瘤?其他?宫颈管少量积液。
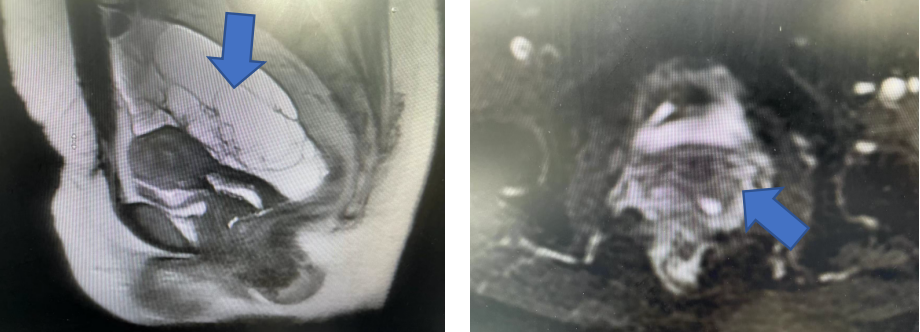
诊疗经过:
患者于2023-01-29行开腹探查术,术中见盆腹腔少量清亮略粘稠液体;子宫形态正常,质软,宫颈未触及增大;双侧卵巢囊性肿物,多囊样,双附件失去正常形态,左侧附件肿物直径15 cm,右侧肿物直径10 cm,均可见输卵管埋藏其中;囊肿表面可见自发破裂口,大网膜与肿物粘连。经腹行扩大子宫切除+双附件切除术,术中左附件送冰冻病理检查,结果提示卵巢黏液性肿瘤,局部上皮生长活跃,建议石蜡充分取材,并建议探查消化道及阑尾。切除大网膜。术中剖视子宫,肉眼观宫颈未见明显异常。
术后病理检查:
(全子宫)宫颈胃型腺癌,癌组织浸润宫颈间质(>1/3且<2/3宫颈壁厚),癌组织向上累及子宫内膜及子宫浅肌层(<1/2肌壁厚),未见确切脉管癌栓及神经侵犯,部分区可见小叶状宫颈腺体增生(LEGH)及非典型小叶状宫颈腺体增生(ALEGH);宫颈周围组织及双宫旁未见癌;子宫平滑肌瘤,局部生长较话跃;可见子宫内膜息肉,其内腺体呈不伴细胞非典的子宫内膜增生症。请结合临床,必要时行分子检测除外P-J综合征(Peutz-Jephersc syndrom)(患者因经济问题暂未进行)。
免疫组化检查:
(宫颈):ER(-),PR(-),P16(-),P53(野生型),Ki-67(局部约30%+),CEA(-),CK20(-),CK7(+),HIKI083(+),MUC6(+),P63(-),PAX2(-),PAX8(+),SATB2(-),Vimentin(-),HLH1(+),MSH2(+),MSH6(+),PMS2(+),特殊染色AB-PAS(+)。(子宫内膜):PAX-2(+),PTEN(+)。特殊染色AB-PAS(-)。(左附件)卵巢黏液性肿瘤,输卵管黏膜可见黏液性肿瘤累及,符合宫颈胃型腺癌转移至卵巢及输卵管。
(左卵巢囊肿):ER(-),PR(-),p53(野生型),ki-67(局部约20%+),WT-1(-),CK7(+),CK20(-),HIK1083(+),MUC6(+),CA125(-),PAX-8(+)。特殊染色AB-PAS(+)。(大网膜)网膜组织未见恶性,可见间皮增生;(右附件)输卵管黏膜可见黏液性肿瘤累及,结合上述情况,符合宫颈胃型腺癌转移至输卵管及卵巢。
术后患者补充放疗+全身化疗中,同时建议患者基因检测。
二、有关Peutz-Jeghers综合征
Peutz-Jeghers综合征(PJS)又称黑斑息肉综合征,是一种以胃肠道息肉病、粘膜皮肤色素斑和某些癌症易感性为特征的遗传综合征,其发病相对罕见,约1/250000~1/300000,我国的发病率约为1/140000,PJS女性患者中发生子宫颈胃型腺癌(Gastric-type endocervical adenocarcinomas,G-EAC)的比例为15%~30%,约50%的G-EAC与PJS有关。该患者已经确诊G-EAC,从组织病理学角度可疑PJS,需要进一步行相关分子检测以明确诊断。为此,我们查阅文献,深入学习PJS的遗传学特征、诊断标准、临床表现及随诊检测,以便结合家族遗传情况,早发现、早诊断及早治疗,并进行长期管理。
01 PJS遗传学特征
PJS是一种常染色体显性遗传病,大多病例是由于STK11基因(也称LKB1基因)胚系突变引起;也有研究发现,脆性组氨酸三联体基因(FHIT基因)可能也参与了PJS疾病的发生。STK11基因位于19号染色体短臂13.3区,基因全长23 kb,由9个编码外显子和1个非编码外显子组成,编码肿瘤抑制因子丝氨酸/苏氨酸激酶。STK11/LKB1参与多种控制细胞周期和细胞增殖的信号通路,包括STK11-AMP活化蛋白-mTOR通路、Wnt信号系统、P53通路及肠上皮细胞中上皮-间充质转化等。
PJS患者中STK11/LKB1突变率约为75%~94%,目前人类基因突变数据库中已确定400余种不同的STK11基因纯合子和复杂的杂合子突变,多集中于1、5、6和7号外显子,突变类型包括无义突变、错义突变、框移突变、剪切突变及大片段缺失,大多数检测到的突变是错义或框移突变,还有30%的突变可能是大片段缺失,仅通过测序技术无法识别。约20%~40%的PJS患者没有家族史且STK11/LKB1未检测到突变。
02 PJS的诊断标准
PJS的诊断基于临床表现、家族史及遗传分析。当满足以下标准之一时,即可诊断为PJS。
-
2个或2个以上病理证实的PJS相关性息肉;
-
任何PJS相关性息肉及PJS家族病史;
-
有PJS家族史的口腔、嘴唇、鼻子、眼睛、生殖器或手指的特征性粘膜皮肤色素沉着;
-
伴有特征性粘膜皮肤色素沉着的PJS相关性息肉。
此外,基因检测、详细的临床病史(包括童年的详细信息)、家族史在诊断中起着关键作用。对确诊PJS病例的亲属应该进行基因检测。
03 PJS患者的临床表现
1、皮肤粘膜色素沉着
约95%的PJS患者出现皮肤黏膜色素沉着,多于幼年期出现, 典型的色素沉着直径1~5 mm, 颜色为深褐色,多分布于眼、耳、口周、肛周、颊黏膜及四肢末端,部分色素沉着可随患者年龄增长而褪色,但口腔粘膜色素沉着终生携带。
2、消化道多发息肉
PJS息肉可在肠外发现,但最常见于胃肠道,小肠息肉约占60%~90%,尤其是空肠;大肠息肉占50%~64%,胃息肉占15%~30%。PJS息肉具有非常典型的组织学特征,其上皮细胞细长、叶状,囊性纤体扩张至粘膜下层或固有基层,且具有“假侵袭”现象(由于平滑肌向上皮细胞延伸导致上皮细胞内陷),此现象在小肠息肉中更为常见,可能误诊为浸润性癌。1/3的PJS患者在10岁时出现息肉症状,1/2在20岁时出现,多以肠套叠、肠梗阻及消化道出血为临床表现。到10岁时,肠套叠风险为15%,20岁时风险增加到50%。
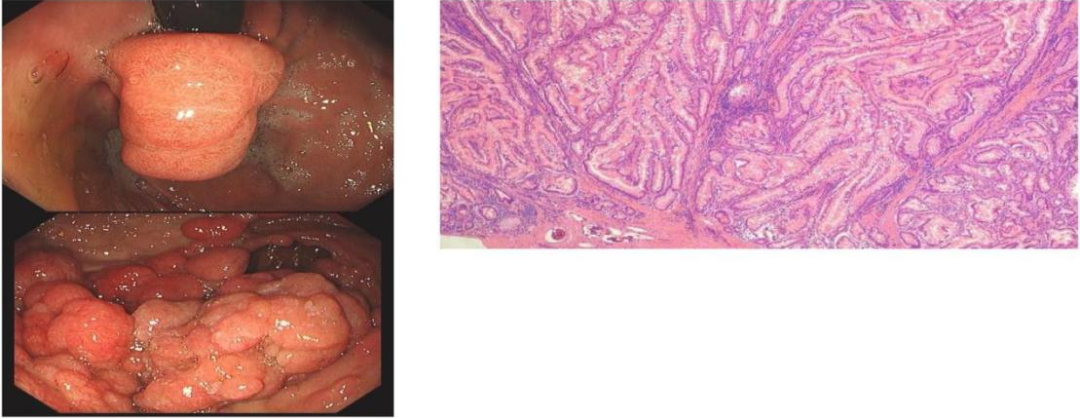
3、肿瘤易感性
PJS患者不仅罹患消化道肿瘤风险高,如结肠直肠癌、胰腺癌和胃癌,患其他非消化道上皮性肿瘤风险也有增加,如乳腺癌、子宫癌、宫颈癌、肺癌、卵巢癌和睾丸癌。在各种恶性肿瘤中,结直肠癌最常见,其次是女性乳腺癌。有研究认为,PJS患者终生罹患恶性肿瘤的风险为37%~93%,平均发病年龄42岁,最常见结直肠癌,终生患病风险达39%,其次是乳腺癌(患病风险32%~54%)、胃癌、小肠癌及胰腺癌;卵巢性索间质肿瘤、宫颈癌及男孩的睾丸支持细胞肿瘤也可在PJS患者中发生。
已有研究发现,PJS女性患妇科肿瘤和乳腺癌的风险增加20.3%,且女性PJS患者的患癌风险显著高于男性,主要原因在于女性罹患妇科恶性肿瘤风险与消化道肿瘤风险相当,甚至高于乳腺癌的患病风险。因此,在肿瘤筛查过程中,妇科恶性肿瘤的筛查应和胃肠道肿瘤同样重视。
PJS患者卵巢中发现性索肿瘤伴环形管(SCTAT),这些患者可表现为月经不调、雌激素增高或性早熟。PJS患者罹患宫颈胃型腺癌,其发病年龄较早,平均年龄33岁,诊断困难且预后差,50%的患者宫颈细胞学筛查结果正常,中位无进展生存约10个月,中位总生存约26个月。
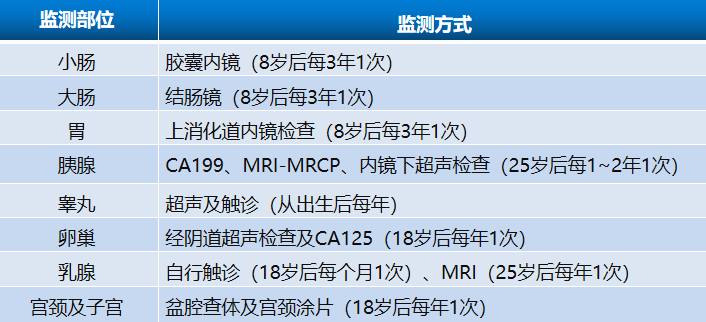
04 随访与监测
对于明确诊断PJS的患者,严密的随访及监测可及早发现胃肠道息肉,并积极治疗预防并发症发生,还可以实现肿瘤的早期诊断,改善预后。有家族史的患者,建议尽早明确基因状态以辅助诊断,具体随访及监测方案见下表。
PJS作为一种遗传性疾病,目前没有有效的治疗措施,结合家族史、临床表现及基因检测,尽早诊断,明确诊断后规范的监测及积极治疗才是改善生活质量及生存的有效途径。
参考文献
[1] Wu M, Krishnamurthy K. Peutz-Jeghers Syndrome [M]. StatPearls Publishing [Internet], 2023.
[2] 石月, 陈奕清, 丁景新等. 中国Peutz-Jeghers综合征女性患者妇科疾病诊治现状调查[J]. 中国癌症杂志, 2022,11: 1049-1064.
[3] 毛旭燕, 张亚飞, 王海丰等. Peutz-Jeghers综合征患者STK11及FHIT基因突变分析[J]. 中华医学遗传学杂志, 2016,02: 186-190.
[4] 张同真, 肖年军, 宁守斌等. Peutz-Jeghers综合征临床及遗传学研究现状[J]. 医学综述, 2021,11: 2233-2238.
[5] 李蒙, 孙涛, 蒋宇亮等. 64例Peutz-Jeghers综合征患者基因变异分析[J]. 中华医学遗传学杂志, 2019,09: 862-863-864-865.
[6] 王文莉, 叶红. Peutz-Jeghers综合征相关宫颈胃型腺癌的研究进展[J]. 中国微创外科杂志, 2022,02: 167-169.
[7] Klimkowski S, Ibrahim M, Ibarra Rovira JJ, et al. Peutz-Jeghers Syndrome and the Role of Imaging: Pathophysiology, Diagnosis, and Associated Cancers. Cancers (Basel). 2021, 13(20): 5121.
[8] Tacheci I, Kopacova M, Bures J. Peutz-Jeghers syndrome [J]. Curr Opin Gastroenterol. 2021, 37(3): 245-254.
[9] Latchford AR, Clark SK. Gastrointestinal aspects of Peutz-Jeghers syndrome [J]. Best Pract Res Clin Gastroenterol. 2022,58-59: 101789.
[10] Tavusbay C, Acar T, Kar H, et al. The patients with Peutz-Jeghers syndrome have a high risk of developing cancer [J]. Turk J Surg. 2018, 34(2):162-164.
[11] Sreemantula HS, Joseph CA, Jamal F, et al. Intussusception Caused by Peutz-Jeghers Syndrome [J]. Cureus. 2022,14(4): e23792.
[12] Li LJ, Wang ZQ, Wu BP. Peutz-Jeghers syndrome with small intestinal malignancy and cervical carcinoma [J]. World J Gastroenterol. 2008, 14(48): 7397-9.
[13] Dos Santos VM, Dos Santos LAM, Modesto LC. Peutz-Jeghers syndrome: revisited. Autops Case Rep. 2022,12: e2021384.
[14] Gordhandas SB, Kahn R, Sassine D, et al, Gastric-type adenocarcinoma of the cervix in patients with Peutz-Jeghers syndrome: a systematic review of the literature with proposed screening guidelines [J]. Int J Gynecol Cancer. 2022, 32(1): 79-88.
[15] Choudhury S, Das A, Misra P, et al. Peutz-Jeghers Syndrome: A Circumventable Emergency [J]. Indian J Dermatol. 2018,63(2): 168-171.

