一次检测、药物任选、不再“一药一伴随”——FDA提出全新的伴随诊断药物审批模式
时间:2023-01-03 15:02:17 热度:37.1℃ 作者:网络
伴随诊断检测(companion diagnostic,CDx)作为一种体外诊断技术,已逐渐成为明确肿瘤患者的生物标志物,保证药物安全有效、提高患者获益的重要方法。目前,肿瘤伴随诊断试剂通常与其获批的特定抗癌药物捆绑在一起,以一种"一药一伴随"的模式出现。但这种模式与患者实际需求之间依然存在很多矛盾。
近日,在美国食品药品监督管理局(FDA)的一次论坛上,FDA肿瘤药中心的Richard Pazdur博士表示,FDA正在试点一项新的伴随诊断审批模式,提出最低性能标准(minimal performance criteria)概念,即针对产品宣称可检测靶点的最低要求,并计划将其嵌入药物临床试验中,然后根据这些标准来进行患者入组。该举措的巨大创新突破在于:药物上市后,医生在临床实践中可直接遵循已公开的靶点检测标准,在市场上选择包含该靶点的检测产品,不必局限于特定的伴随诊断检测产品。“一药一伴随”的固有方式即将被打破。
在肿瘤靶向药物飞速发展的十年间,无论是国家药品监督管理局(NMPA)还是FDA等医疗器械监管机构,都在不断思考、不断探索更优的伴随诊断开发模式。例如,FDA在明确伴随诊断定义之后,于2016年至2020年间逐步完善对伴随诊断试剂的监管。与此同时,我国也在不断规范伴随诊断产品的管理。
作为肿瘤精准治疗的重要环节之一,NMPA在2020年发布了《已上市抗肿瘤药物的伴随诊断试剂临床试验指导原则(征求意见稿)》,定义了抗肿瘤药伴随诊断的适用范围和开发标准。2022年6月,局医疗器械技术审评中心(CDME) 发布了《与肿瘤药物同步研发的原研伴随诊断试剂临床试验注册审查指导原则》, 进一步指导抗肿瘤药的原研伴随诊断试剂上市前所需临床试验和注册申报资料准备。新的指导原则根据已上市产品是否满足伴随诊断需求,将伴随诊断的开发分为共同开发(co-development)和桥接(bridging study)等方式。
在共同开发模式下,一款伴随诊断试剂从开发、完成性能验证和临床试验验证、到最终与药物同步上市要经历至少3-5年时间。上市后,药物和伴随诊断试剂一一对应,并在药物或试剂标签中作相应的提示。但这种"一药一伴随"的模式仍然存在很多现实临床问题。
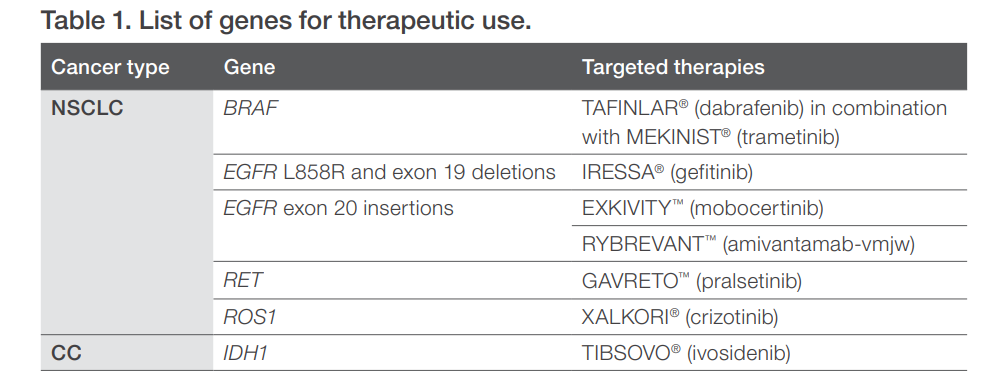
例如,EGFR 20ins是近两年晚期非小细胞肺癌中备受关注的罕见靶点,2021年先后有两款药物获得FDA批准,分别是武田制药的莫博替尼胶囊(Exkvity,TAK-788) 和强生的埃万妥单抗(Rybrevant,,amivantamab) 注射液。同时获批的是两款药物的伴随诊断试剂,分别为赛默飞的Oncomine Dx作为莫博替尼的伴随诊断,以及Guardant Health的Guardant360作为埃万妥单抗的伴随诊断。如果患者需要从莫博替尼更换为埃万妥单抗,则需要重新进行对应伴随诊断检测,即使这两款药物是针对的是同一靶点。这样的多次检测意味着患者进行检测的样本量需求、时间周期和花销随之翻倍。此外,医院对检测平台的选择、药物可及性等问题,也成为横亘在患者与顺利用药之间的难题。
在规划的伴随诊断产品需求更新的情况下,桥接试验是最快能够满足产品升级的方法。桥接试验听似简单,实则暗藏挑战。桥接实验使用临床试验的剩余样本,用预期进行目标试剂重新检测,比较两种试剂之间的性能,因此对剩余样本的管理、保藏、溯源、人类遗传资源审批、运输等各个环节都需要严格把控,以确保样本的有效性和合规性。这些都是伴随诊断企业进行桥接试验必须投入的精力和成本。即使是成熟的伴随诊断试剂,完成整个过程到最终上市也可能需要1年左右的时间。例如上文提到的埃万妥单抗,FDA在埃万妥单抗上市半年多之后(2021年12月)才根据桥接试验的结果,批准Oncomine Dx也可以用于其诊断。对于亟需更换药物的患者来说,时间就是生命,桥接试验可能仍然无法伴随诊断快速上市的临床需求。
OncomineDx伴随诊断基因
FDA的新提案希望进一步优化现有模式,加速伴随诊断审批,让伴随诊断更好更快地服务临床患者。根据最低性能标准概念,随着药物的获批上市,只要符合用药标准的伴随诊断检测产品的结果都可以作为相关靶点药物的依据,从一对一变成一对多,甚至多对多,大大扩展了医生和患者对伴随诊断产品的选择。患者的样本和时间都弥足珍贵,这一方式不仅提高样本利用率,减少重复采样带来的痛苦,也可以为患者节省下更多检测成本。
对于进行伴随诊断试剂的企业来说,只要检测符合最低性能标准,即可广泛适用所有同类靶向药物。节约企业在共同开发或桥接实验过程中针对每款药物进行性能验证、开展临床试验的成本,缩短试剂的开发周期;这一新模式的提出,进而可以给患者提供更加平价的检测试剂,实现良性循环。
在肿瘤精准医学领域快速发展的时代,尤其是NGS多基因检测的优势逐渐得到认可之际,漫长的注册审批程序可能导致伴随诊断产品不能快速适应新指南的速度,无法更好地服务于临床。FDA新草案的提出或可使肿瘤伴随诊断试剂的应用场景更加灵活,最大化患者获益、降低医疗决策难度、控制企业投产比。合理参考借鉴这一新方法,有助于提高国内伴随诊断产品在国际赛道上的竞争力。
·END ·
: , 。 视频 小程序 赞 ,轻点两下取消赞 在看 ,轻点两下取消在看

