各类肾病接招!WES测序助力遗传诊断率超1/3,一些人甚至改变临床诊断和治疗
时间:2023-12-12 10:38:53 热度:37.1℃ 作者:网络
通过对172例不同肾脏疾病的儿童或成人患者的DNA进行全外显子组测序(WES),评估遗传性肾脏疾病(GKD)的基因谱,以及遗传诊断在患者治疗中的应用。在63例(36.6%)患者中,WES诊断出遗传性疾病。肾小球疾病患者中共检出10个基因变异,诊断率为33.8%(25/74);肾小管间质疾病患者中共检出18个基因变异,诊断率为58.8%(20/34);囊性疾病/纤毛病患者中共检出10个基因变异,诊断率为33.3%(15/45);先天性肾脏和尿路异常(CAKUT)患者中共检出2个基因变异,诊断率为18.2%(2/11);终末期肾病(ESKD)的患者诊断率为12.5%(1/8)。<1- 6岁年龄组的确诊率高(46-50.0%),≥40岁年龄组的确诊率低(9.1%)。遗传诊断后,63例患者中,有10例(15.9%)患者的肾脏表型被重新分类,10例(15.9%)患者的临床治疗发生改变。综上所述,这些发现证明了WES的诊断效用及其在不同年龄组的各种肾脏疾病患者中的有效临床应用。
研究背景
遗传性肾脏病(GKD)是一种罕见的疾病,约有10%的成人终末期肾脏病(ESKD)患者存在GKD。相比之下,GKD在<25岁的肾病患者中占很大比例(高达70%)。迄今为止,超过200个单基因或染色体结构异常已被证实可导致<25岁患者的慢性肾脏病(CKD)。这一遗传复杂性使患者的诊断变得复杂,其中有相当大比例的患者未得到诊断。
大规模并行基因组测序技术的发展,如全外显子组测序(WES),提高了诊断过程的速度和效率,特别是对于具有高基因组异质性的单基因变异的患者。迄今为止,WES已在9.3%-40.0%的病因不明CKD或家族性肾病患者中发现了遗传原因。WES对先天性肾脏和尿路异常(CAKUT)的患者诊断率为5%,对肾结石/肾钙质沉着症的患者诊断率为11%-20.8%,对激素耐药型肾病综合征的患者诊断率为30%,对Alport综合征的患者诊断率为55%-80%,对纤毛病型肾小管肥大症的患者诊断率为21%-25%。
然而,迄今为止,很少有研究评估WES在不同肾脏疾病患者中的诊断率及其临床应用。因此,本研究评估了WES在韩国各种肾脏疾病患者队列中的诊断率。此外,评估WES对临床的影响,如遗传确诊后的疾病重新分类、患者管理和家庭成员咨询的变化。
研究结果
临床特征:
2018年4月至2021年11月,共有172例患者(男性88例,女性84例)被转诊至韩国首尔峨山医疗中心医学遗传学中心,以评估可能的潜在遗传性肾脏疾病。172例患者均来自170个无血缘关系的韩国家庭。4例患者来自两个无亲缘关系家系,其中一个家系:父亲和女儿因家族性肾小球病疑有蛋白尿,但未发现显著的致病变异;另1个家系:母子疑似常染色体显性肾小管间质疾病(ADTKD)所致青少年型高尿酸血症肾病,未发现相关致病变异。患者的人口统计学特征见表1。在患者被纳入本研究时,共有97例(56.3%,97/172)中位年龄为12.5岁(范围0.0-73.0)的患者已经出现肾功能不全(评估的GFR<90mL/min/m2)。在这些患者中,43例(25%)有肾脏疾病家族史。发病的中位年龄为7.8岁(范围0-71.9岁),其中发病年龄<10岁者105例(61.0%),10-20岁者44例(25.6%),20-40岁者12例(7.0%),>40岁者11例(6.4%)。基因检测的中位年龄为11.6岁(范围0.2-73岁)。纳入时的临床诊断包括74例(43.0%)肾小球病、34例(19.8%)的肾小管间质疾病、45例(26.2%)肾囊性疾病/纤毛病、11例(6.4%)CAKUT和8例(4.7%)原因不明的ESKD(表2)。每个年龄组的表型和疾病谱可能不同。分层时,12-18岁肾小球病患者最多(30/172,17.4%),其次为1岁的囊性疾病/纤毛病(16/172,9.3%)和18-40岁肾小球病(16/172,9.3%)。由于按年龄和表型分类的各亚型患者数量太少,未进行进一步的亚组分析。
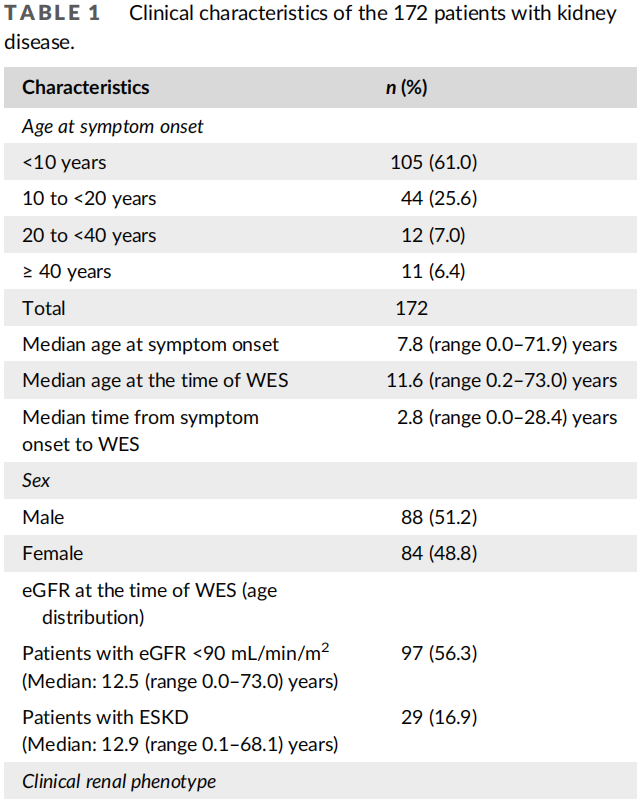

表1
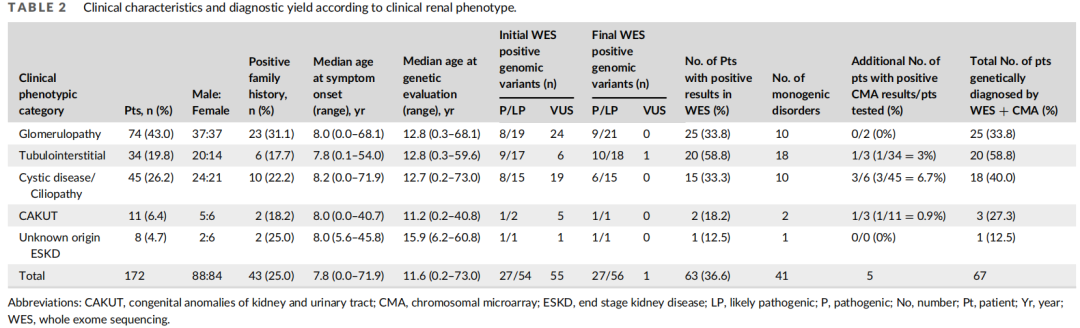
表2
基于WES的遗传诊断:
172例患者均行WES检测。根据ACMG指南,最小变异丰度>5%的变异,以及可能是良性、良性和证据较少的非编码变异被过滤掉,只选择在符合每例患者表型的基因中发现的变异。阳性结果为致病性(P)和可能致病性(LP)变异。对于具有不确定意义的变异(VUS)患者,当家族成员检测发现新生现象(de novo phenomenon)以常染色体显性方式遗传,或当家族成员检测发现复合杂合以常染色体隐性方式遗传,计算机模拟分析显示明显的功能恶化时,研究者认为结果为阳性。最初,WES在16例患者中发现了27个P变异,在47例患者中发现了55个LP变异,在46例患者中发现了55个VUS变异。除1个变异外,对所有这些变异进行的Sanger测序均获得了相同的结果,随后对80例患者进行了家族成员检测。2个LP变异由于不匹配的遗传模式、临床表型和缺乏用于验证的家族检测而被排除。1例患者(表3中的患者14)携带两个复合杂合变异,分别为P和VUS,根据ACMG规则,并在计算机分析、明显的表型关系和家庭分析的支持下得到了确诊。最后,63例患者的84个变异(包括27个P变异、56个LP变异和1个VUS变异)被认为是阳性结果(图1),即在172例患者中,63例(36.6%)得到遗传学诊断。在63例患者中,4例(表3中的第1、2、11和17例)已由作者描述。不同临床表型的变异致病性分布见表2。

图1
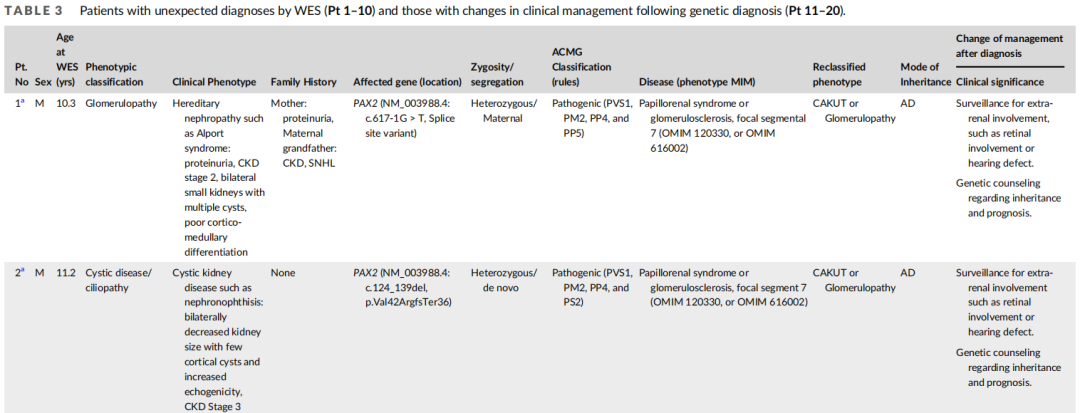







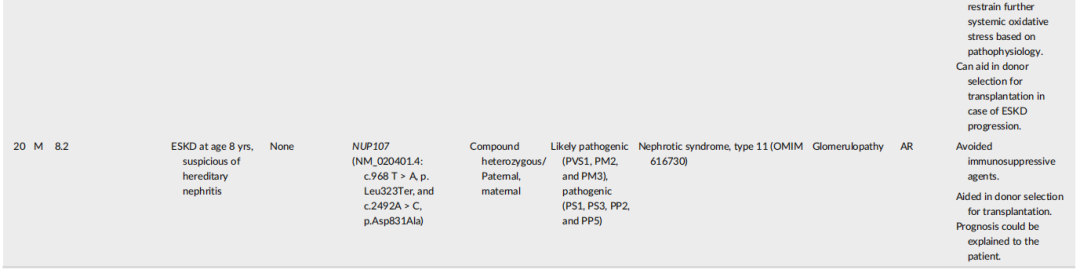
表3
基于发病年龄和肾脏表型的WES诊断率:
从症状出现到遗传评估的中位间隔时间为2.8年(范围0-28.4年)。在年龄<1岁(46.2%,18/39)、1-3岁(50.0%,7/14)和3-6岁(46.7%,7/15)的患者中诊断率较高,在6-12岁(35.2%,19/54)、8-40岁(28.6%,4/14)和12-18岁(28.0%,7/25)的患者中诊断率较低。在年龄≥40岁的患者中诊断率最低(9.1%,1/11)(p=0.232,图2)。研究者还评估了肾脏表型和诊断率之间的关联(表2和图3)。肾小管间质疾病患者的诊断率最高(58.8%,20/34),其次是肾小球病(33.8%,25/74)、囊性疾病/纤毛病(33.3%,15/45)和CAKUT(18.2%,2/11),但这些差异无统计学意义(p>0.05)。总体而言,80.9%(51/63)的诊断变异经通过Sanger测序进行的其他家族检测证实,包括21例肾小球病患者、16例肾小管间质疾病患者、11例囊性疾病/纤毛病患者、2例CAKUT患者和1例病因不明ESKD患者(图3)。

图2
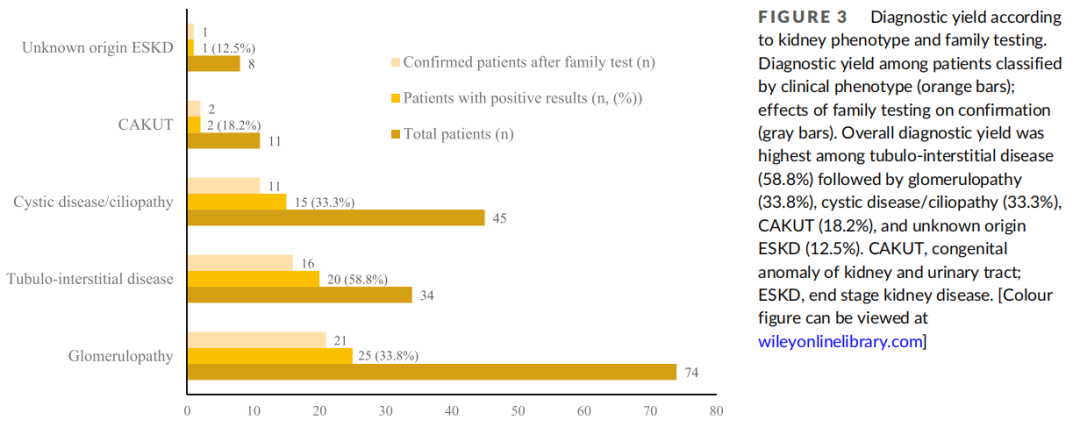
图3
基于肾脏表型的基因变异谱:
各表型的基因谱见图4。25例肾小球疾病患者共检出10种单基因病,最常见的为COL4A5(48.0%,12/25),其次为NUP107(12.0%,3/25)、INF2(8.0%,2/25)、WT1(8.0%,2/25)、COL4A3(4.0%,1/25)、COQ8(4.0%,1/25)、CUBN(4.0%,1/25)、MAF(4.0%,1/25)、NPHS1(4.0%,1/25)和PAX2(4.0%,1/25)。在20例肾小管间质疾病患者中共检出18种单基因疾病,其中SLC12A3和UMOD变异各见于2例患者(11%)。该组的其余患者在剩余15个基因中各有1个不同的基因变异:ACE、AGXT、APRT、CDKN1C、CUBN、CYP24A1、GRHPR、HNF1B、HOGA1、HPRT1、KIF11、PHEX、SLC12A1、SLC22A12、SLC26A3和TRPM6。15例囊性病/纤毛病患者中共检出10种单基因病。最常见的变异基因为PKD1(33.3%,5/15),其次为PKHD1(13.3%,2/15)、BUB1B(6.7%,1/15)、CSPP1(6.7%,1/15)、NPHP3(6.7%,1/15)、LRP2(6.7%,1/15)、PAX2(6.7%,1/15)、PDHA1(6.7%,1/15)、TSC2(6.7%,1/15)和ZEB2(6.7%,1/15)。在2例CAKUT表型的ESKD患者中分别检测到ZEB2和PAX2基因变异,NPHP3基因变异见于1例病因不明的ESKD患者。
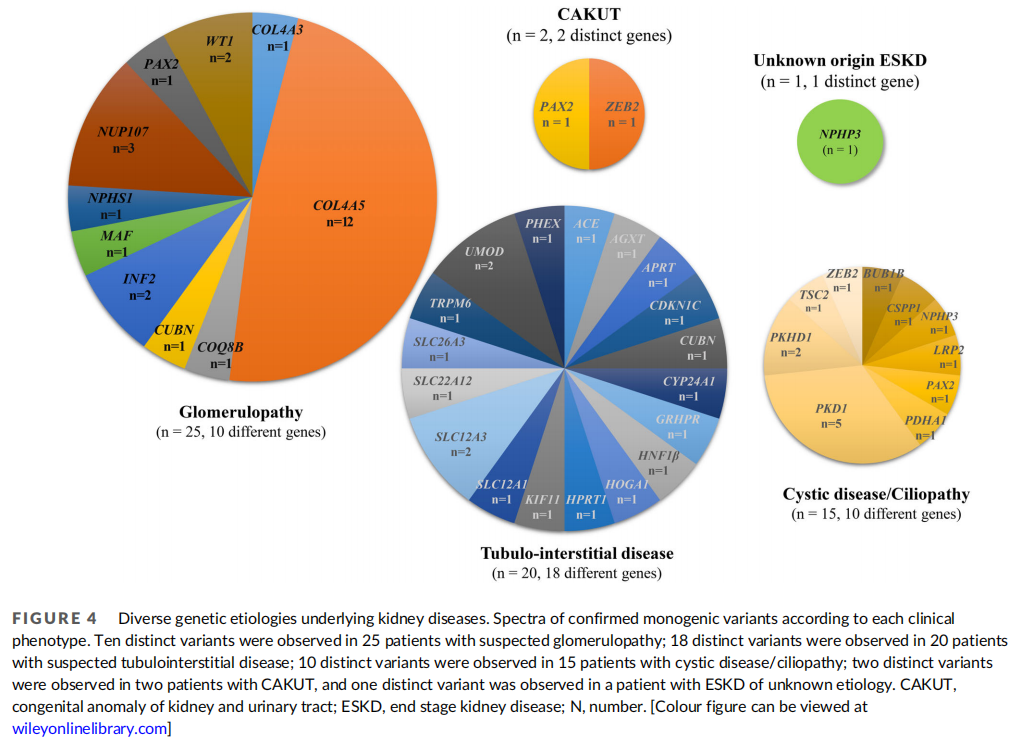
图4
遗传诊断后GKD的重新分类:
在63例遗传诊断的患者中,53例(84%)的临床诊断与遗传诊断一致,包括23例肾小球病、18例肾小管间质疾病、10例囊性/纤毛病和2例CAKUT。其余10例根据遗传学结果重新分类,包括1例从肾小球病重新分类为CAKUT(Pt1),1例从肾小球病重新分类为肾小管间质疾病(Pt4),2例从肾小管间质疾病重新分类为CAKUT(Pt5、Pt10),1例从囊性疾病/纤毛病重新分类为肾小管间质疾病(Pt6),以及1例从病因不明的ESKD重新分类为囊性疾病/纤毛病(Pt3)(表3)。其余3例患者由囊性疾病/纤毛病重新分类为CAKUT(Pt 2、7、9),1例患者由囊性疾病/纤毛病重新分类为肾小管间质疾病或肾小球病(Pt 8)。6例患者(Pt 1- 6)可根据不同的表型分类进行诊断,4例疑似综合征性疾病患者(Pt 7-Pt 10)可明确综合征特征的病因。
遗传诊断对GKD管理的临床影响:
遗传诊断改变了63例遗传诊断患者中10例(15.9%)患者的临床管理(Pt 11-20,表3)。在5例复发性肾结石患者中,2例分别被诊断为PRT和HPRT1缺陷的患者(Pt 11和12)接受了黄嘌呤氧化酶抑制剂治疗,以防止结石形成和肾功能不全进展。1例诊断为AGXT缺陷的患者(Pt 13)是lumasiran治疗的候选患者,一种在肾移植后通过靶向乙醇酸氧化酶来减少肝草酸生成的RNA干扰剂。另2例(Pt 14和Pt 15)分别诊断为GRHPR缺陷和HOGA1缺陷的患者,给予枸橼酸钾治疗。Pt 16的CUBN变异使其被诊断为慢性良性蛋白尿(OMIM 618884),从而避免了有创肾活检或免疫抑制剂治疗。Pt 17因TRPM6变异被诊断为肠道低镁血症1型(OMIM 602014),开始补充镁。在Pt 18、19和20中分别发现NUP107、NPHS1和COQ8B变异,使其停止免疫抑制剂治疗,而在Pt 19中发现的COQ8B变异,表明需要补充辅酶Q10。
WES和管理的次要发现:
根据ACMG指南,2例患者出现了意外的次要发现。1例因PKD1基因变异疑似患有囊性肾病的女性患者也携带LP RYR1基因变异。根据ACMG指南和致病性评估,对该家系进行恶性高血压的风险咨询。1例因NUP107基因变异而患有肾小球疾病的男性患者同时携带LP MYH11基因变异,该变异遗传自母亲。该变异与发生动脉瘤或主动脉夹层或主动脉瓣病变(主动脉瘤,家族性胸椎4;OMIM 132900)。因此,建议该患者及其母亲定期接受心功能评估。
染色体微阵列(CMA)分析的额外诊断:
14例患者行CMA分析,包括1例基于WES检测疑似携带CNV,和13例合并多发先天畸形及发育迟缓。CMA分析显示5例患者有5种诊断,3例患者为17q12缺失(包括HNF1β),包括1例肾钙质沉着症、多发肾囊肿、低镁血症和青少年的成人型糖尿病(MODY)患者, 2例为具有综合征疾病特征和发育落后的先天性囊性肾病患者。此外,检测到1例双侧肾脏发育不良伴皮质囊肿、面容异常、听力下降的3月龄男童存在16q11q12微缺失,和在一名有鲜红痣、单侧多囊性肾发育不良、先天性心脏病、发育迟缓、畸形、白内障的7月龄女童中检测到一个46,XX, der(21)t(6;21) (p25;q22.1)mat [20]/46, XX[50]基因型(表2)。
讨 论
本研究评估了WES在儿童和成人各种类型肾脏疾病患者中的效用以及该方法的临床适用性。本研究的结果是有意义的,因为描述了各种肾脏疾病的整体基因谱,并提示了根据年龄组和肾脏亚表型的不同的遗传诊断率。本研究的总体诊断率为36.6%。相比之下,WES之前对病因不明的CKD或家族性肾病患者的诊断率为9.3%-60.0%,诊断率取决于患者的年龄、种族和近亲比例,以及纳入患者的数量和纳入标准、临床表型的定义/分类和代表性表型的同质性分级。本研究中WES对儿童GKD的诊断率与既往对儿童GKD的诊断率(36.2%-42.6%)相当。
在本研究的172例患者中,<20岁患者有135例(78.5%),WES在该年龄段的诊断率最高(40.7%,55/135)。相比之下,成人的诊断率为29.7%(11/37)。最近的研究还表明,就诊时年龄较小与较高的诊断率独立相关,这表明早期进行基因检测的重要性。在本研究中,从症状出现到基因检测的中位时间为2.8(0-28.4)年,需要进一步缩短这一间隔。虽然成年起病患者的总体诊断率略低,但遗传诊断具有重要的临床意义。如Pt 13,肾移植后肾结石反复发作,肾功能不全进行性加重,遗传诊断为APRT缺陷,提示需要降尿酸治疗以防止进一步的肾损伤。
在患者人群存在的各种肾脏疾病中,WES对肾小管间质疾病的诊断率最高(58.8%)。在既往研究中,WES在该表型中也显示出较高的诊断率(61.1%-100%)。这一高诊断率可能与这些患者的临床和生化特征相关,包括围生期病史、生长迟缓和特异性电解质失衡伴酸碱失衡。此外,本研究中82%的患者发病年龄为<20岁。肾小球病和囊性病/纤毛病的诊断率为33%-34%,在有阳性家族史的患者中诊断率分别为提升至40.9%和50.0%。在本研究中,WES对主要包括激素耐药型肾病综合征/FSGS和Alport综合征的肾小球疾病患者的诊断率与既往研究相当(7.2%-55.8%)。
常染色体显性多囊肾病(ADPKD)是最常见的囊性疾病/纤毛病,主要通过直接测序法确诊。由于重叠的同源区域和高GC含量使得该基因难以被WES捕获,因此通过WES进行诊断受到了关注。但在疑似ADPKD患者中,使用定制探针进行WES的变异检出率与直接测序相当,检出率高达83.3%-99.0%。在一项研究中,WES对囊性肾病患者的总体诊断率为27%,其中>90%的患者由于PKD1和PKD2的变异而患ADPKD。本研究未纳入通过直接测序诊断为ADPKD的患者,在确诊为囊性疾病/纤毛病的患者中,只有33%患ADPKD。因此,在这些囊性疾病/纤毛病患者中33.3%的诊断率应被认为与以往研究的诊断率相当或更高。
WES对先证者及其父母(三人)的诊断率高于既往研究中先证者单独应用WES的诊断率(44.8% vs. 36.2%)。然而,在实际临床实践中,由于费用和可及性,仅先证者测序更常被选择。本研究仅对先证者进行测序,随后对家系成员的DNA进行Sanger测序,以确认先证者发现的变异的致病性。这些方法的诊断率相当。
本研究中各疾病组的总体基因谱与既往研究相似。肾小球病一般可分为两个亚组,一组是遗传性肾炎,48%的患者有COL4A5变异,4%有COL4A3变异;另一组为蛋白尿,36%的患者患有以NUP107、INF2、WT1、NPHS1和COQ8变异为特征的单基因足细胞病。与之前的研究相似,本研究发现COL4A5变异是肾炎的主要病因。本研究中足细胞病的基因谱也与之前对WT1、NPHS1、NUP107、COQ6和COQ8变异导致的韩国激素抵抗型肾病综合征患者的研究结果一致。这一基因谱与白种人不同,白种人的变异发生率较高,尤其是在NPHS2、PLCE1、LAMB2和TRPC6中。其他研究也观察到种族差异,这表明足细胞病在东亚人群中有独特的基因谱,在中国、日本和韩国患者中,WT1、NPHS1、INF2和COQ8的变异发生率较高。
在本研究中,肾小管间质疾病表现出最不均匀的肾脏疾病分布。这些亚型包括与通道病相关的遗传性肾小管病,主要是Gitleman或Bartter综合征;常染色体显性遗传肾小管间质疾病(ADTKD)谱,以UMOD为主;以及其他引起肾结石的原因。肾小管间质疾病的异质性在之前的队列研究中也有报道,这些研究不仅包括CKD患者,还包括各种GKD表型的患者。例如,CLCN5、SLC12A3、SLC4A1、AGXT、OCRL、SLC12A1和ATP6V1B1在中国疑似肾小管疾病患儿中均有发现。对澳大利亚一个队列进行的分析显示,成人携带UMOD、FAN1、ATP6V1B1和CASR变异,儿童携带KCNJ1、CLCNKB、CLCN5、SLC12A3、CLDN16、SLC34A1和AVPR2变异。本研究中的囊性疾病/纤毛病的特征主要是PKD1变异,然后是PKHD1变异,而不是既往关于成人囊性肾病的研究中报道的PKD2变异。超过70%有这一表型的患者年龄在20岁以下,80%的确诊病例属于这一年龄组。本研究参与者的年龄分布和临床诊断为ADPKD的患者比例低可能部分解释了疾病的分布,正如在其他纳入儿童的研究中观察到的那样。分布于各个临床表型组的表型包括肾小球病和囊性疾病/纤毛病中的PAX2变异,以及肾小管间质疾病中的HNF1β变异。
基于WES结果提示的一些疾病是出乎意料的,15.9%(10/63)的确诊变异患者进行了疾病重新分类。同样,在既往研究中,20-40%的患者在基因分析后需要重新分类。WES等基因分析后的这种相反表型分型对于遗传病的诊断确认至关重要,并且可能有助于理解某一基因型的表型异质性。此外,遗传诊断以及肾脏或肾外受累的确定,可使医师根据每个基因型量身定制治疗,从而提高患者的治疗效果。如果没有遗传诊断,这种定制方案将是困难的。例如,在Pt 7检出提示CAKUT的ZEB2变异,并伴有阴茎阴囊转位和全面发育,因此进一步评估了该患者的心脏异常、听力丧失和巨结肠引起的肠扩张,并计划对肾功能和尿液分析进行评估,因为该患者可能表现为肾小球囊性疾病。
遗传诊断可以使临床医生更好地了解疾病的病理生理和管理。准确的诊断可以改变患者的治疗。例如,在Pt 17中发现TRPM6变异后,治疗从补钙改为补镁,单基因足细胞病的发现提示应避免使用免疫抑制剂。准确诊断还可以提供新的治疗选择,以减缓疾病进展,例如,携带AGXT变异的Pt 13使用Lumasiran,携带COQ8变异的Pt 19使用COQ10。在63例遗传确诊的疾病患者中,研究者观察到10例(15.9%)患者的临床治疗发生了变化。对于CAKUT或纤毛病患者,应进行肾外系统监测。在63例遗传确诊的患者中,7例(11%)携带COL4A5、NUP107、UMOD和NPHP3变异的患者接受了在世亲属供体选择咨询。最后,研究者与患者及其家庭成员讨论了未来的生育计划,以及建议进行指导监测的次要结果(RYR1、MYH11)。
由于结构变异(CNVs)可能是部分CAKUT疾病的病因,因此CMA可进一步在部分GKD患者中发挥补充诊断作用。在本研究纳入的172例患者中,14例(8.1%)患者进行了CMA,诊断率为35.7%(5/14)。这5例患者中有3例存在17q12微缺失,而大约65%的基因病患者存在6个最常见的位点中的一个变异(1q12.1、4p16.1-16.3、16p11.2、16p13.11、17q12和22q11.2)。因此,对于疑似综合征性疾病伴有包括CAKUT在内的多种异常的病例,即使WES未检出结构变异,也建议进行CMA检查。
本研究有几个局限性。WES本身在识别所有重要的遗传变异方面有固有的局限性,包括存在于非编码内含子/启动子区域的变异、CNVs以及结构变异,包括数量可变的串联重复序列、线粒体变异和覆盖率低的外显子。对于初次WES阴性的患者,应考虑追加CMA。由于进行了index-only WES,因此可能遗漏了一些变异,或者由于缺乏父母和未患病家系成员的数据,致病性评估受到影响。对于常染色体隐性遗传基因变异的致病性,尽管存在复合杂合变异和一致的表型,但VUS的遗传诊断仍存在争议。虽然本研究纳入的患者数量不少,但所有患者均为朝鲜族。因此,这些结果不能推广到其他种族。
本研究结果证明了WES在各种肾脏疾病患者中的诊断效用和有效的临床应用。测序成本的降低和分析技术的进步可能会增强WES在肾脏疾病患者诊断和管理中的临床适用性。
参考文献:
Jung, Jiwon et al. “Genetic diagnosis of kidney disease by whole exome sequencing and its clinical application.” Clinical genetics vol. 104,3 (2023): 298-312. doi:10.1111/cge.14382


