无脉络膜症:症状和体征,病因,流行病学,诊断和治疗
时间:2023-09-25 08:26:05 热度:37.1℃ 作者:网络
无脉络膜症(choroideremia,progressive choroidal atrophy)又称全脉络膜血管萎缩、进行性脉络膜萎缩、进行性毯层脉络膜萎缩。其特点是双眼进行性发病,自幼夜盲,弥漫性全层脉络膜毛细血管及RPE萎缩,最后脉络膜完全消失。与原发性视网膜变性有所不同,此病被认为是脉络膜的缺失。经过长期观察发现脉络膜与色素上皮并不是先天性发育不良而是后天进行性消失,故又称为进行性RPE营养不良性变性或进行性RPE脉络膜变性。
一、症状与体征
无脉络膜血症的特征是眼睛所有视网膜层的广泛丧失。 这种疾病通常始于儿童时期,伴有色素性视网膜上皮、视网膜和脉络膜的消耗(萎缩)。 视网膜是光敏感的最内层,由许多含有神经的层组成。 视网膜外侧有一层单色素细胞。 脉络膜是位于视网膜和眼睛“白色”部分(巩膜)之间的下一层; 该层含有小血管)。
脉络膜血管退化后会导致视网膜损伤,这通常会导致周边视力丧失,最终可能导致失明。 中央视力通常保留到晚年。 无脉络膜血症的症状在受影响的个体之间可能有很大差异。 女性携带者通常有非常轻微的症状,包括晚年出现的夜盲症或对眩光敏感。
1.视功能改变
起病较早,可能出生时已有,视力甚至下降至光感。视野进行性向心性缩小。萎缩改变开始于中周边部,渐成管状视野。通常10~30岁时视力中度下降但仍保持中央视野,40~50岁累及黄斑后,最后视野完全消失,患者完全失明。夜盲早发,晚期则测不出暗适应曲线,色觉紊乱,为红绿色盲眼。
2.眼底改变
眼底改变可出现在婴幼儿期,亦可较晚,甚至40岁以后只有初期的改变。眼底改变可分为3期为初期、中期、晚期。
女性患者为携带者,典型的眼底改变与年轻的男性患者相仿。但眼底病变为静止性,程度轻,且视力正常。眼底可表现为色素脱失及色素增生,呈椒盐状萎缩多位于眼底赤道部,色素颗粒大小不等,向周边排列成串且为放射条状。极周边部则色素减退。有些病例黄斑部有细小的色素沉着视网膜及视盘正常。
二、流行病学
无脉络膜症(choroideremia, CHM)是一种X连锁隐形遗传的视网膜疾病,伴有视网膜色素上皮(RPE)、光感受器、脉络膜的进行性萎缩。男性患者常常在20岁前出现夜盲,随后出现进行性的周边视野缺损,变成管状视野最终变为法定盲人。在晚期,黄斑区也会萎缩并出现中心视野缺损。CHM的患病率在1:50000-100000。
三、病因
无脉络膜症的致病基因是CHM基因,位于Xq21.2,编码蛋白质REP-1。该病的男性患者发病较早,症状从早期的(十几岁~二十几岁)夜盲症逐渐发展为周边视野缺失,到晚年仅存中心管状视野,最终失明,女性携带者一般无症状。
1.遗传因素。本病为X染色体隐性遗传已被公认。男性发病,且为进行性,女性为基因携带者。目前认为是CHM基因是主要原因。
无脉络膜症是以X连锁隐性模式遗传的。 CHM基因位于两条性染色体之一的X染色体上。 在男性(只有一个X染色体)中,每个细胞中一个改变的基因拷贝足以导致该病症。 在女性(有两条X染色体)中,基因的两个拷贝都必须存在突变才能导致疾病。 与女性相比,男性更易患X连锁隐性遗传疾病。 X连锁遗传的一个特征是父亲不能将X连锁特征传递给他们的儿子。 携带CHM突变的女性可能会在视网膜内显示出细小的细胞丢失区域,可在彻底的眼睛检查过程中观察到。 这些改变会在以后的生活中损害视力。
CHM是由于CHM基因突变失活,导致Rep-1缺失或者产生无功能的基因产物,从而出现脉络膜血管层发育障碍,进行性RPE和脉络膜营养不良、变 性及进行性脉络膜萎缩。CHM基因定位于染色体Xq21.2区域,包含15个外显子,跨度约150×103碱基对。该基因编码一个包含653个氨基酸的胞内蛋白质Rabescortprotein-1(Rep -1)。无脉络膜症是一种以视力减退为特征的病症,主要影响男性。 这种情况的首要症状通常是夜间视力受损(夜盲症),这可能发生在儿童早期。 视野逐渐变窄(隧道视野),以及看到细节(视力)的能力下降。 这些视力问题是由于在眼睛后部(视网膜)和附近血管网络(脉络膜)排列的专用光敏组织中细胞(萎缩)的持续损失所致。 随着时间的推移,脉络膜血症的视力损害恶化,但患病进程各不相同。 然而,所有患有这种疾病的人都会出现失明,最常见的是在成年后期。

CHM 基因关联的疾病表型:

CHM患者的突变分布
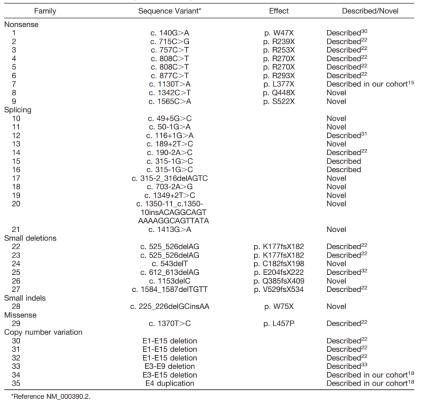
2.免疫因素
从组织学和超微结构中发现有巨噬细胞吞噬色素和光感受器外节,在吞噬的色素中有关蛋白质可能引起过敏反应,使脉络膜间质崩溃、Bruch膜和血管消失从而导致脉络膜全层萎缩。
三、诊断与检查
1、视功能改变 起病较早,可能出生时已有,视力减退甚至下降至光感。视野进行性向心性缩小。萎缩改变开始于中周边部,随着年龄增长向中心扩展,最后残余中央部分,渐成管状视野。通常10~30岁时视力中度下降,但仍保持中央视野,40~50岁累及黄斑后,为管视或伴有周边部小岛,最后视野完全消失。至中心视岛及颞侧残存视岛消失后患者完全失明。夜夜盲早发,呈杆锥型变性,暗适应视杆细胞终阈值呈进行性升高,晚期则测不出暗适应曲线。色觉紊乱,为红绿色盲。眼电生理测量,早期明视ERG正常,暗适应ERG为低波,晚期熄灭。EOG低波或无波。本病男性患者ERG的改变为病变早期ERG明适应部分可正常,但暗适应部分的a波和b波振幅降低,b波潜伏期延长,晚期ERG熄灭。女性患者的视力、视野、暗适应、色素、EOG、ERG多为正常,但偶有异常。女性携带者在眼底呈现明显的蚕食样色素紊乱和堆积的情形下,其ERG反应仍正常,ERG振幅可降低或增高。本病的EOG的改变比ERG更明显,无脉络膜症患者视功能检查的一个异常特征是EOG基线电位的明显下降,晚期EOG基线电位几乎测不到,光峰完全消失。女性患者的视功能多无异常改变,但少数患者可有异常。
2、眼底改变 眼底改变可出现在婴幼儿期,亦可较晚,甚至40岁以后只有初期的改变。可分为3期。
(1)初期:有轻度非典型周边部色素性视网膜病变,由于视网膜色素上皮变性,眼底赤道及周边部呈闪辉黄色,深部有色素颗粒,色素之间有脱色素区,故眼底呈椒盐状。色素不呈骨细胞状。
(2)中期:病变逐渐从周边向后极部发展,视网膜的内层无色素,这时出现脉络膜血管和RPE萎缩,表现小区域的脉络膜大血管暴露。
(3)晚期:脉络膜及RPE向眼底后极部进行性萎缩。眼底的色素上皮几乎完全被破坏,脉络膜血管消失并萎缩,有时黄斑部仅留下一小块脉络膜,其边界清楚。同时周边部可残留一脉络膜小岛,但到50~60岁后亦逐渐消失。由于色素上皮和脉络膜的血管消失,眼底暴露出巩膜的白色反光。残留的小岛可呈棕红色,伴有周边部的圆形或不规则的色素斑,但很少见到全眼底均为白色,无可见的脉络膜血管。虽然脉络膜病变明显,视网膜及视神经常保持正常。晚期视盘可萎缩,视网膜血管可稍细。
女性患者为携带者,典型的眼底改变与年轻的男性患者相仿。但眼底病变为静止性,程度轻,且视力正常。眼底可表现为色素脱失及色素增生,呈椒盐状萎缩,多位于眼底赤道部,色素颗粒大小不等,向周边排列成串且为放射条状。极周边部则色素减退。有些病例黄斑部有细小的色素沉着,视网膜及视盘正常。
3、荧光血管造影 早期RPE缺损呈现广泛的强荧光区,继而视网膜色素上皮萎缩和脉络膜毛细血管消失,仅见荧光充盈的脉络膜大血管。晚期可见广泛的无荧光区,其中残存稀疏的脉络膜大血管。女性基因携带者的荧光造影可见RPE萎缩,呈透见荧光或广泛的强荧光。
本病男性患者的临床特征为夜盲、视野向心性缩窄、蓝色觉异常、暗适应阈值升高及进行性全脉络膜血管和RPE萎缩。根据典型的眼底改变、电生理改变及家族史,可做出正确诊断。
眼底荧光血管造影检查可以辅助诊断。早期RPE缺损呈现广泛的强荧光区继而视网膜色素上皮萎缩和脉络膜毛细血管消失,仅见荧光充盈的脉络膜大血管,晚期可见广泛的无荧光区其中残存稀疏的脉络膜大血管。女性基因携带者的荧光造影可见RPE萎缩呈透见荧光或广泛的强荧光。
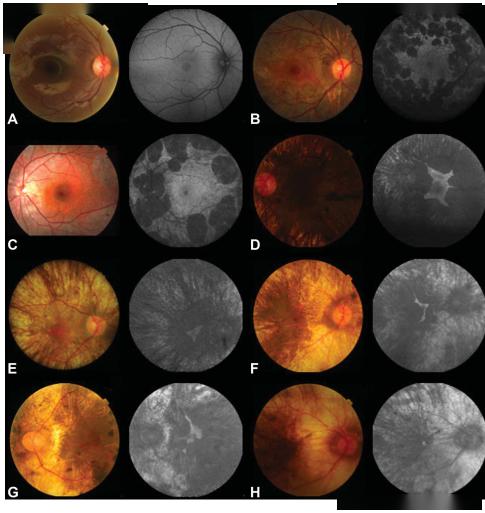
图3. 8个CHM患者的眼底和FAF图片。患者的年龄从A至H分别为6、8、18、24、37、44、52岁。
诊断
本病男性患者的临床特征为夜盲,视野向心性缩窄,蓝色觉异常,暗适应阈值升高及进行性全脉络膜血管和RPE萎缩,根据典型的眼底改变,电生理改变及家族史,可做出正确诊断。
四、鉴别诊断
(1)视网膜色素变性
无脉络膜症的早期与RP鉴别困哪,在实际临床工作中,我们也很少诊断早期的无脉络膜症病例,这些病例估计多被误诊为RP了。无脉络膜症的中期,虽然在眼底可见到类似于视网膜变性的色素上皮萎缩性改变,但无典型的骨细胞样色素、血管相对正常和无视盘的改变有助于两者的鉴别。另外,与性连锁的RP相比,无脉络膜症的黄斑功能保持时间相对较长。无脉络膜症的晚期眼底比较典型,有助于鉴别。
(2)回旋状脉络膜萎缩
回旋状脉络膜萎缩和无脉络膜症这两种疾病在早期均容易误诊为视网膜变性,在晚期眼底改变基本相同,仅凭眼底检查无法鉴别。鉴别诊断主要依靠无脉络膜症的阳性家族史、回旋状脉络膜萎缩中期的典型眼底改变、回旋状脉络膜萎缩的化验检查结果(存在高鸟氨酸血症和多种体液中鸟氨酸水平明显升高)。
(3)弥漫性脉络膜毛细血管萎缩 仅限于RPE及脉络膜毛细血管层,荧光血管造影明显可见。其遗传特点为常染色体显性遗传。
(4)白化病眼底 有明显的白化病表现,不伴有夜盲及视野改变。
(5)病理性近视 不仅有高度近视,而且有巩膜葡萄肿等有改变。
五、治疗
1、标准治疗
无特殊治疗方法。
2、临床试验
目前无脉络膜症无有效的药物治疗,随着技术的进展,国外已开展1/2 期基因治疗临床实验。基因治疗目的是重获细胞内的蛋白运输、减少视网膜细胞内代谢产物的累积,从而减慢或终止视力的下降。在许多视网膜萎缩性疾病的基因治疗中,重组腺相关病毒2(AAV2)被证明对光感受器细胞和RPE具有良好的亲和力,因此作为常用的基因载体被用于治疗相关视网膜疾病。无脉络膜症基因治疗是利用AAV2-REP1载体将编码REP1的CHM 基因运送到RPE层并在视网膜光感受器细胞中表达,已有大量数据证明AAV2视网膜下给药的安全性,具有良好的治疗前景。
目前,研究人员采用一种新的基因治疗方法,使这种疾病患者的视力得以恢复。这项技术,利用单次注射,将眼部的一个有缺陷基因,替换为这个基因的正常工作拷贝。 这项I期试验的研究结果,发表在2014年1月16日的《柳叶刀》(The Lancet)杂志上,支持在更常见遗传因素造成的失明(包括老年退行性疾病如黄斑变性,和遗传缺陷如色素性视网膜炎)中,进一步开发这种最先进的治疗方法。
MacLaren及其同事们,在35-63岁之间、处于无脉络膜症不同阶段的6位患者中,评估了基因疗法对视网膜和视功能的治疗效果。他们将一个载体——一种经基因方法改造的腺相关病毒(adeno-associated virus,AAV),注入患者视网膜,将这个基因的矫正版本,传递到眼部的适当部位,从而终止感光细胞的死亡。 这种疗法没有造成伤害,使视觉的主观测量得到好转。在基因传递6个月后,所有患者都恢复了视力,两位患者表现出实质性的视力改善。重要的是,与未治疗的眼睛灵敏度丧失相比,患者的感光敏感度增加。
六、预防
本症病程可迁延数年,急性发作及缓解交替出现,大多数到成年期自行缓解。但也有少数仍持续发作。如关节炎多年不愈,可造成严重关节畸形,活动障碍。此多见于多关节炎型,发病年龄较大的女孩及全身型伴有多关节炎者。少关节炎型,在4岁前发病的女孩,多发生慢性虹膜睫状体炎,造成失明。强直性脊椎炎可见于发病年龄较大的男孩。总的来说,如能及时治疗,75%患者病情缓解,关节功能正常。只有少数造成终身残废。个别患者合并感染或淀粉样变性而死亡。
七、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
罕见疾病患者信息登记系统

